

Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI
- PMID: 11381124
- PMCID: PMC34700
- DOI: 10.1073/pnas.121027598
Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI
Abstract
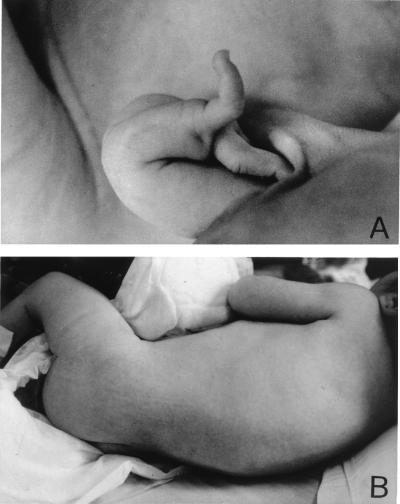
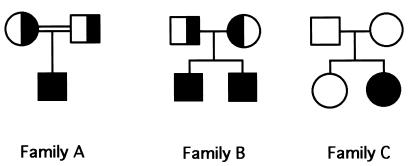
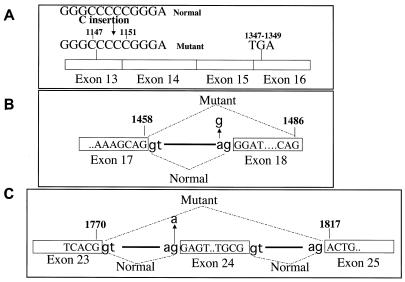
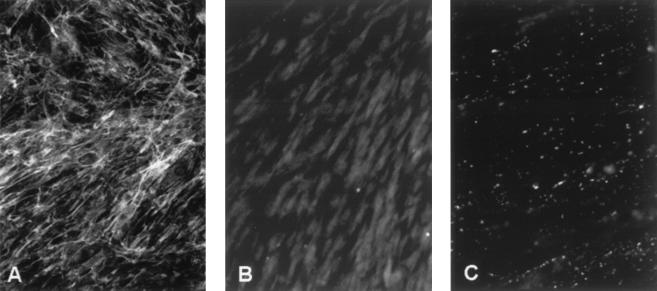
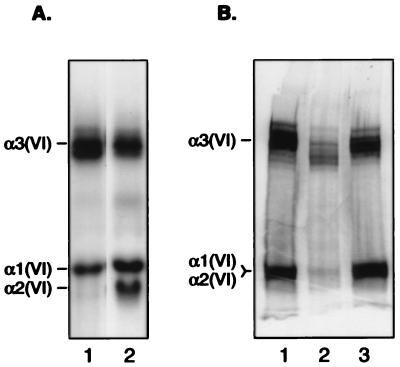
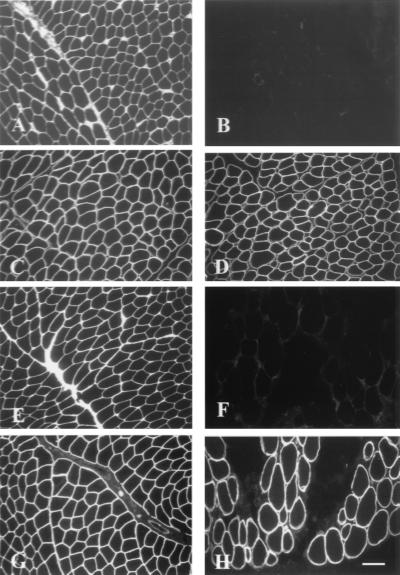
Ullrich syndrome is a recessive congenital muscular dystrophy affecting connective tissue and muscle. The molecular basis is unknown. Reverse transcription-PCR amplification performed on RNA extracted from fibroblasts or muscle of three Ullrich patients followed by heteroduplex analysis displayed heteroduplexes in one of the three genes coding for collagen type VI (COL6). In patient A, we detected a homozygous insertion of a C leading to a premature termination codon in the triple-helical domain of COL6A2 mRNA. Both healthy consanguineous parents were carriers. In patient B, we found a deletion of 28 nucleotides because of an A --> G substitution at nucleotide -2 of intron 17 causing the activation of a cryptic acceptor site inside exon 18. The second mutation was an exon skipping because of a G --> A substitution at nucleotide -1 of intron 23. Both mutations are present in an affected brother. The first mutation is also present in the healthy mother, whereas the second mutation is carried by their healthy father. In patient C, we found only one mutation so far-the same deletion of 28 nucleotides found in patient B. In this case, it was a de novo mutation, as it is absent in her parents. mRNA and protein analysis of patient B showed very low amounts of COL6A2 mRNA and of COL6. A near total absence of COL6 was demonstrated by immunofluorescence in fibroblasts and muscle. Our results demonstrate that Ullrich syndrome is caused by recessive mutations leading to a severe reduction of COL6.
Figures
Similar articles
-
Dominant and recessive COL6A1 mutations in Ullrich scleroatonic muscular dystrophy.Ann Neurol. 2005 Sep;58(3):400-10. doi: 10.1002/ana.20586. Ann Neurol. 2005. PMID: 16130093
-
Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy.Am J Hum Genet. 2002 Jun;70(6):1446-58. doi: 10.1086/340608. Epub 2002 Apr 24. Am J Hum Genet. 2002. PMID: 11992252 Free PMC article.
-
Identification and characterization of novel collagen VI non-canonical splicing mutations causing Ullrich congenital muscular dystrophy.Hum Mutat. 2009 May;30(5):E662-72. doi: 10.1002/humu.21022. Hum Mutat. 2009. PMID: 19309692
-
Collagen type VI myopathies.Adv Exp Med Biol. 2014;802:185-99. doi: 10.1007/978-94-007-7893-1_12. Adv Exp Med Biol. 2014. PMID: 24443028 Review.
-
[Collagen VI-related muscle disorders].Brain Nerve. 2011 Nov;63(11):1169-78. Brain Nerve. 2011. PMID: 22068469 Review. Japanese.
Cited by
-
Animal models of muscular dystrophy.Prog Mol Biol Transl Sci. 2012;105:83-111. doi: 10.1016/B978-0-12-394596-9.00004-4. Prog Mol Biol Transl Sci. 2012. PMID: 22137430 Free PMC article. Review.
-
Endotrophin, a Key Marker and Driver for Fibroinflammatory Disease.Endocr Rev. 2024 May 7;45(3):361-378. doi: 10.1210/endrev/bnad036. Endocr Rev. 2024. PMID: 38091968 Free PMC article. Review.
-
Natural history of pulmonary function in collagen VI-related myopathies.Brain. 2013 Dec;136(Pt 12):3625-33. doi: 10.1093/brain/awt284. Epub 2013 Nov 22. Brain. 2013. PMID: 24271325 Free PMC article.
-
From proteins to genes: immunoanalysis in the diagnosis of muscular dystrophies.Skelet Muscle. 2011 Jun 24;1(1):24. doi: 10.1186/2044-5040-1-24. Skelet Muscle. 2011. PMID: 21798100 Free PMC article.
-
A Nonsense Variant in COL6A1 in Landseer Dogs with Muscular Dystrophy.G3 (Bethesda). 2015 Oct 4;5(12):2611-7. doi: 10.1534/g3.115.021923. G3 (Bethesda). 2015. PMID: 26438297 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
