

Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome
- PMID: 11170895
- PMCID: PMC1235280
- DOI: 10.1086/318201
Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome
Abstract
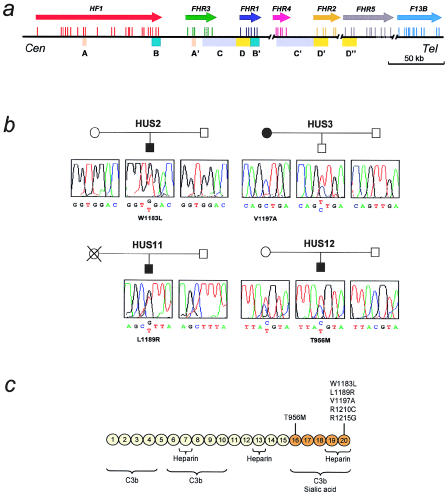
Hemolytic-uremic syndrome (HUS) is a microvasculature disorder leading to microangiopathic hemolytic anemia, thrombocytopenia, and acute renal failure. Most cases of HUS are associated with epidemics of diarrhea caused by verocytotoxin-producing bacteria, but atypical cases of HUS not associated with diarrhea (aHUS) also occur. Early studies describing the association of aHUS with deficiencies of factor H suggested a role for this complement regulator in aHUS. Molecular evidence of factor H involvement in aHUS was first provided by Warwicker et al., who demonstrated that aHUS segregated with the chromosome 1q region containing the factor H gene (HF1) and who identified a mutation in HF1 in a case of familial aHUS with normal levels of factor H. We have performed the mutational screening of the HF1 gene in a novel series of 13 Spanish patients with aHUS who present normal complement profiles and whose plasma levels of factor H are, with one exception, within the normal range. These studies have resulted in the identification of five novel HF1 mutations in four of the patients. Allele HF1 Delta exon2, a genomic deletion of exon 2, produces a null HF1 allele and results in plasma levels of factor H that are 50% of normal. T956M, W1183L, L1189R, and V1197A are missense mutations that alter amino acid residues in the C-terminal portion of factor H, within a region--SCR16-SCR20--that is involved in the binding to solid-phase C3b and to negatively charged cellular structures. This remarkable clustering of mutations in HF1 suggests that a specific dysfunction in the protection of cellular surfaces by factor H is a major pathogenic condition underlying aHUS.
Figures
Similar articles
-
Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome.Am J Hum Genet. 2002 Dec;71(6):1285-95. doi: 10.1086/344515. Epub 2002 Nov 6. Am J Hum Genet. 2002. PMID: 12424708 Free PMC article.
-
Complete factor H deficiency-associated atypical hemolytic uremic syndrome in a neonate.Pediatr Nephrol. 2007 Jun;22(6):874-80. doi: 10.1007/s00467-007-0438-x. Epub 2007 Feb 13. Pediatr Nephrol. 2007. PMID: 17295030
-
Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease.Hum Mol Genet. 2003 Dec 15;12(24):3385-95. doi: 10.1093/hmg/ddg363. Epub 2003 Oct 28. Hum Mol Genet. 2003. PMID: 14583443
-
Atypical hemolytic uremic syndrome.Orphanet J Rare Dis. 2011 Sep 8;6:60. doi: 10.1186/1750-1172-6-60. Orphanet J Rare Dis. 2011. PMID: 21902819 Free PMC article. Review.
-
Implications of the initial mutations in membrane cofactor protein (MCP; CD46) leading to atypical hemolytic uremic syndrome.Mol Immunol. 2007 Jan;44(1-3):111-22. doi: 10.1016/j.molimm.2006.07.004. Epub 2006 Aug 1. Mol Immunol. 2007. PMID: 16882452 Review.
Cited by
-
Kidney Disease Caused by Dysregulation of the Complement Alternative Pathway: An Etiologic Approach.J Am Soc Nephrol. 2015 Dec;26(12):2917-29. doi: 10.1681/ASN.2015020184. Epub 2015 Jul 16. J Am Soc Nephrol. 2015. PMID: 26185203 Free PMC article. Review.
-
Analysis of rare variants in the CFH gene in patients with the cuticular drusen subtype of age-related macular degeneration.Mol Vis. 2015 Mar 15;21:285-92. eCollection 2015. Mol Vis. 2015. PMID: 25814826 Free PMC article.
-
The disease-protective complement factor H allotypic variant Ile62 shows increased binding affinity for C3b and enhanced cofactor activity.Hum Mol Genet. 2009 Sep 15;18(18):3452-61. doi: 10.1093/hmg/ddp289. Epub 2009 Jun 23. Hum Mol Genet. 2009. PMID: 19549636 Free PMC article.
-
The molecular and structural bases for the association of complement C3 mutations with atypical hemolytic uremic syndrome.Mol Immunol. 2015 Aug;66(2):263-73. doi: 10.1016/j.molimm.2015.03.248. Epub 2015 Apr 11. Mol Immunol. 2015. PMID: 25879158 Free PMC article.
-
Genetic and environmental factors influencing the human factor H plasma levels.Immunogenetics. 2004 May;56(2):77-82. doi: 10.1007/s00251-004-0660-7. Epub 2004 Apr 30. Immunogenetics. 2004. PMID: 15118848
References
Electronic-Database Information
-
- Entrez Nucleotide Sequence Search, http://www.ncbi.nlm.nih.gov/Entrez/nucleotide.html (for factor H cDNA sequence [accession number Y00716] and genomic sequences of the HF1 gene and the FHR1–FHR5 genes [accession numbers AL049744, AL049741, AL139418, and AL353809])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for autosomal dominant [MIM 134370] and recessive [MIM 235400] HUS)
References
-
- Ault BH (2000) Factor H and the pathogenesis of renal diseases. Pediatr Nephrol 14:1045–1053 - PubMed
-
- Ault BH, Schmidt BZ, Fowler NL, Kashtan CE, Ahmed AE, Vogt BA, Colten HR (1997) Human factor H deficiency: mutations in framework cysteine residues and block in H protein secretion and intracellular catabolism. J Biol Chem 272:25168–25175 - PubMed
-
- Blackmore TK, Hellwage J, Sadlon TA, Higgs N, Zipfel PF, Ward HM, Gordon DL (1998) Identification of the second heparin-binding domain in human complement factor H. J Immunol 160:3342–3348 - PubMed
-
- Blackmore TK, Sadlon TA, Ward HM, Lublin DM, Gordon DL (1996) Identification of a heparin binding domain in the seventh short consensus repeat of complement factor H. J Immunol 157:5422–5427 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
