

Paternal origin of FGFR2 mutations in sporadic cases of Crouzon syndrome and Pfeiffer syndrome
- PMID: 10712195
- PMCID: PMC1288162
- DOI: 10.1086/302831
Paternal origin of FGFR2 mutations in sporadic cases of Crouzon syndrome and Pfeiffer syndrome
Abstract
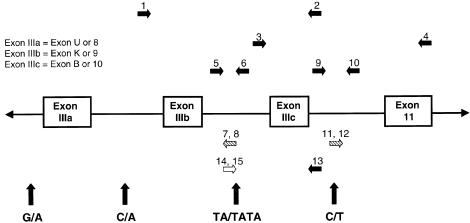
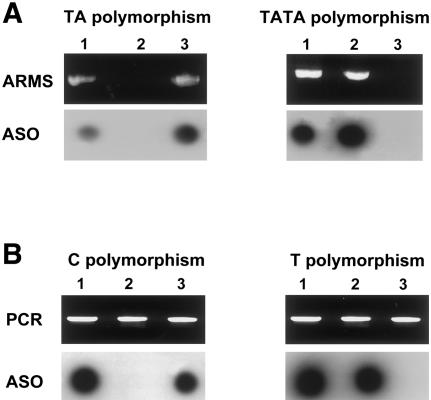
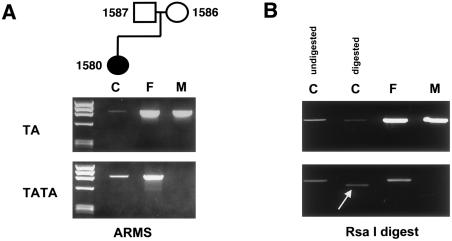
Crouzon syndrome and Pfeiffer syndrome are both autosomal dominant craniosynostotic disorders that can be caused by mutations in the fibroblast growth factor receptor 2 (FGFR2) gene. To determine the parental origin of these FGFR2 mutations, the amplification refractory mutation system (ARMS) was used. ARMS PCR primers were developed to recognize polymorphisms that could distinguish maternal and paternal alleles. A total of 4,374 bases between introns IIIa and 11 of the FGFR2 gene were sequenced and were assayed by heteroduplex analysis, to identify polymorphisms. Two polymorphisms (1333TA/TATA and 2710 C/T) were found and were used with two previously described polymorphisms, to screen a total of 41 families. Twenty-two of these families were shown to be informative (11 for Crouzon syndrome and 11 for Pfeiffer syndrome). Eleven different mutations in the 22 families were detected by either restriction digest or allele-specific oligonucleotide hybridization of ARMS PCR products. We molecularly proved the origin of these different mutations to be paternal for all informative cases analyzed (P=2. 4x10-7; 95% confidence limits 87%-100%). Advanced paternal age was noted for the fathers of patients with Crouzon syndrome or Pfeiffer syndrome, compared with the fathers of control individuals (34. 50+/-7.65 years vs. 30.45+/-1.28 years, P<.01). Our data on advanced paternal age corroborates and extends previous clinical evidence based on statistical analyses as well as additional reports of advanced paternal age associated with paternal origin of three sporadic mutations causing Apert syndrome (FGFR2) and achondroplasia (FGFR3). Our results suggest that older men either have accumulated or are more susceptible to a variety of germline mutations.
Figures

Similar articles
-
FGFR2 exon IIIa and IIIc mutations in Crouzon, Jackson-Weiss, and Pfeiffer syndromes: evidence for missense changes, insertions, and a deletion due to alternative RNA splicing.Am J Hum Genet. 1996 Mar;58(3):491-8. Am J Hum Genet. 1996. PMID: 8644708 Free PMC article.
-
Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes.Nat Genet. 1995 Feb;9(2):173-6. doi: 10.1038/ng0295-173. Nat Genet. 1995. PMID: 7719345
-
Description of a new mutation and characterization of FGFR1, FGFR2, and FGFR3 mutations among Brazilian patients with syndromic craniosynostoses.Am J Med Genet. 1998 Jul 7;78(3):237-41. Am J Med Genet. 1998. PMID: 9677057
-
[From gene to disease; craniosynostosis syndromes due to FGFR2-mutation].Ned Tijdschr Geneeskd. 2002 Jan 12;146(2):63-6. Ned Tijdschr Geneeskd. 2002. PMID: 11820058 Review. Dutch.
-
Clinicogenetic study of Turkish patients with syndromic craniosynostosis and literature review.Pediatr Neurol. 2014 May;50(5):482-90. doi: 10.1016/j.pediatrneurol.2014.01.023. Epub 2014 Jan 11. Pediatr Neurol. 2014. PMID: 24656465 Review.
Cited by
-
Paternal factors and schizophrenia risk: de novo mutations and imprinting.Schizophr Bull. 2001;27(3):379-93. doi: 10.1093/oxfordjournals.schbul.a006882. Schizophr Bull. 2001. PMID: 11596842 Free PMC article. Review.
-
Reproductive genetics and the aging male.J Assist Reprod Genet. 2018 Jun;35(6):933-941. doi: 10.1007/s10815-018-1148-y. Epub 2018 Mar 9. J Assist Reprod Genet. 2018. PMID: 29524155 Free PMC article. Review.
-
Fibroblast Growth Factor Receptor 2 (FGFR2) Mutation Related Syndromic Craniosynostosis.Int J Biol Sci. 2017 Nov 2;13(12):1479-1488. doi: 10.7150/ijbs.22373. eCollection 2017. Int J Biol Sci. 2017. PMID: 29230096 Free PMC article. Review.
-
SMAD4 mutations causing Myhre syndrome are under positive selection in the male germline.Am J Hum Genet. 2024 Sep 5;111(9):1953-1969. doi: 10.1016/j.ajhg.2024.07.006. Epub 2024 Aug 7. Am J Hum Genet. 2024. PMID: 39116879 Free PMC article.
-
Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease.Am J Hum Genet. 2012 Feb 10;90(2):175-200. doi: 10.1016/j.ajhg.2011.12.017. Am J Hum Genet. 2012. PMID: 22325359 Free PMC article. Review.
References
Electronic-Database Information
-
- CEPH, http://www.cephb.fr/
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for oligonucleotide primers and nucleotide numbers [accession number AF169399])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for Crouzon syndrome [MIM 123500] and Pfeiffer syndrome [MIM 101600])
References
-
- al-Qattan MM, Phillips JH (1997) Clinical features of Crouzon's syndrome patients with and without a positive family history of Crouzon's syndrome. J Craniofac Surg 8:11–13 - PubMed
-
- Cohen MM Jr (1993) Pfeiffer syndrome update, clinical subtypes, and guidelines for differential diagnosis. Am J Med Genet 45:300–307 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
