

Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots
- PMID: 10577905
- PMCID: PMC1288362
- DOI: 10.1086/302690
Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots
Abstract
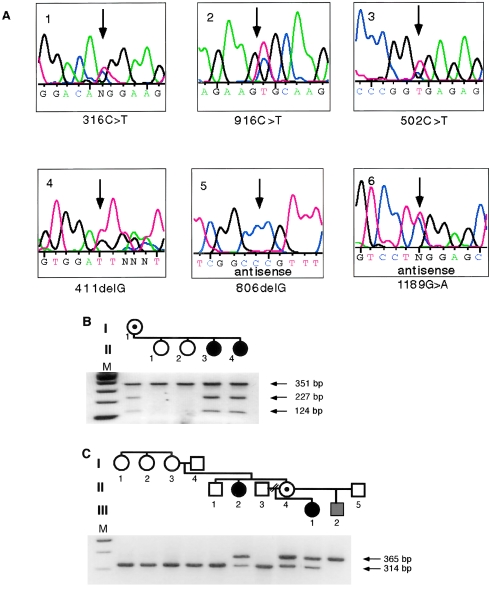
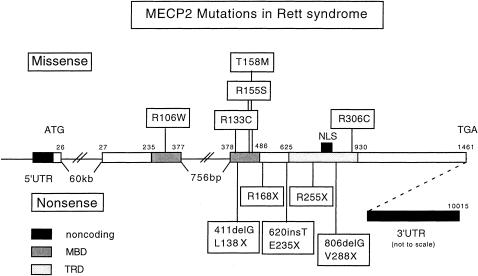
Rett syndrome (RTT) is a neurodevelopmental disorder characterized by loss of acquired skills after a period of normal development in infant girls. The responsible gene, encoding methyl-CpG binding protein 2 (MeCP2), was recently discovered. Here we explore the spectrum of phenotypes resulting from MECP2 mutations. Both nonsense (R168X and R255X) and missense (R106W and R306C) mutations have been found, with multiple recurrences. R168X mutations were identified in six unrelated sporadic cases, as well as in two affected sisters and their normal mother. The missense mutations were de novo and affect conserved domains of MeCP2. All of the nucleotide substitutions involve C-->T transitions at CpG hotspots. A single nucleotide deletion, at codon 137, that creates a L138X stop codon within the methyl-binding domain was found in an individual with features of RTT and incontinentia pigmenti. An 806delG deletion causing a V288X stop in the transcription-repression domain was identified in a woman with motor-coordination problems, mild learning disability, and skewed X inactivation; in her sister and daughter, who were affected with classic RTT; and in her hemizygous son, who died from congenital encephalopathy. Thus, some males with RTT-causing MECP2 mutations may survive to birth, and female heterozygotes with favorably skewed X-inactivation patterns may have little or no involvement. Therefore, MECP2 mutations are not limited to RTT and may be implicated in a much broader phenotypic spectrum.
Figures
Similar articles
-
Diagnostic testing for Rett syndrome by DHPLC and direct sequencing analysis of the MECP2 gene: identification of several novel mutations and polymorphisms.Am J Hum Genet. 2000 Dec;67(6):1428-36. doi: 10.1086/316913. Epub 2000 Oct 30. Am J Hum Genet. 2000. PMID: 11055898 Free PMC article.
-
Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2.Nat Genet. 1999 Oct;23(2):185-8. doi: 10.1038/13810. Nat Genet. 1999. PMID: 10508514
-
Rett syndrome: the complex nature of a monogenic disease.J Mol Med (Berl). 2003 Jun;81(6):346-54. doi: 10.1007/s00109-003-0444-9. Epub 2003 May 16. J Mol Med (Berl). 2003. PMID: 12750821 Review.
-
Rett syndrome: methyl-CpG-binding protein 2 mutations and phenotype-genotype correlations.Am J Med Genet. 2000 Summer;97(2):147-52. doi: 10.1002/1096-8628(200022)97:2<147::aid-ajmg6>3.0.co;2-o. Am J Med Genet. 2000. PMID: 11180222 Review.
-
Functional consequences of Rett syndrome mutations on human MeCP2.Nucleic Acids Res. 2000 Nov 1;28(21):4172-9. doi: 10.1093/nar/28.21.4172. Nucleic Acids Res. 2000. PMID: 11058114 Free PMC article.
Cited by
-
Comprehensive High-Depth Proteomic Analysis of Plasma Extracellular Vesicles Containing Preparations in Rett Syndrome.Biomedicines. 2024 Sep 24;12(10):2172. doi: 10.3390/biomedicines12102172. Biomedicines. 2024. PMID: 39457485 Free PMC article.
-
Genetic Analysis of MECP2 Gene in Iranian Patients with Rett Syndrome.Iran J Child Neurol. 2019 Summer;13(3):25-34. Iran J Child Neurol. 2019. PMID: 31327966 Free PMC article.
-
Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain.Proc Natl Acad Sci U S A. 2002 Nov 26;99(24):15536-41. doi: 10.1073/pnas.242566899. Epub 2002 Nov 13. Proc Natl Acad Sci U S A. 2002. PMID: 12432090 Free PMC article.
-
Functional diversification of the nematode mbd2/3 gene between Pristionchus pacificus and Caenorhabditis elegans.BMC Genet. 2007 Aug 28;8:57. doi: 10.1186/1471-2156-8-57. BMC Genet. 2007. PMID: 17725827 Free PMC article.
-
Rett syndrome and MeCP2: linking epigenetics and neuronal function.Am J Hum Genet. 2002 Dec;71(6):1259-72. doi: 10.1086/345360. Epub 2002 Nov 19. Am J Hum Genet. 2002. PMID: 12442230 Free PMC article. Review. No abstract available.
References
-
- Amir RE, Roth Dahle E, Toniolo D, Zoghbi HY. Candidate gene analysis in Rett syndrome and the identification of twenty-one SNPs in Xq. Am J Med Genet (in press) - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
