

Clinical follow-up of 2 families with glomerulopathy caused by COQ8B gene variants and literature review
- PMID: 39877334
- PMCID: PMC11772357
- DOI: 10.3389/fped.2024.1378083
Clinical follow-up of 2 families with glomerulopathy caused by COQ8B gene variants and literature review
Abstract
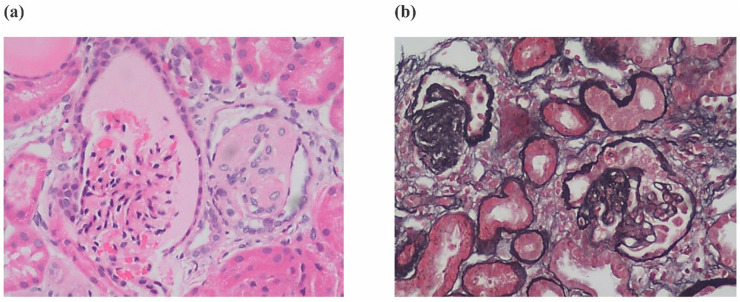
Background: Primary coenzyme Q10 (CoQ10) deficiency is an autosomal recessive genetic disease caused by mitochondrial dysfunction. Variants in Coenzyme Q8B (COQ8B) can cause primary CoQ10 deficiency. COQ8B-related glomerulopathy is a recently recognized glomerular disease that most often presents as steroid-resistant nephrotic syndrome (SRNS) in childhood. The disease often progresses to kidney failure and the renal histopathology is most commonly focal segmental glomerulosclerosis (FSGS).
Methods: Four SRNS cases (2 females and 2 males) from 2 unrelated families who were followed clinically for nearly 3 years. Clinical exome testing and analyses were performed by MyGenostics Laboratory in China to evaluate unexplained proteinuria given the strong family history of glomerular disease and histologic evidence of SRNS. Pathogenic variants were identified in COQ8B in the exome studies and confirmed by direct sequencing.
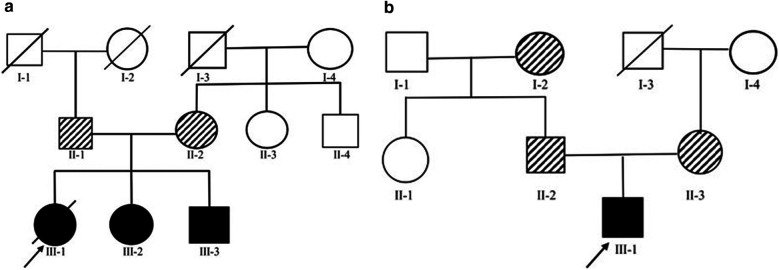
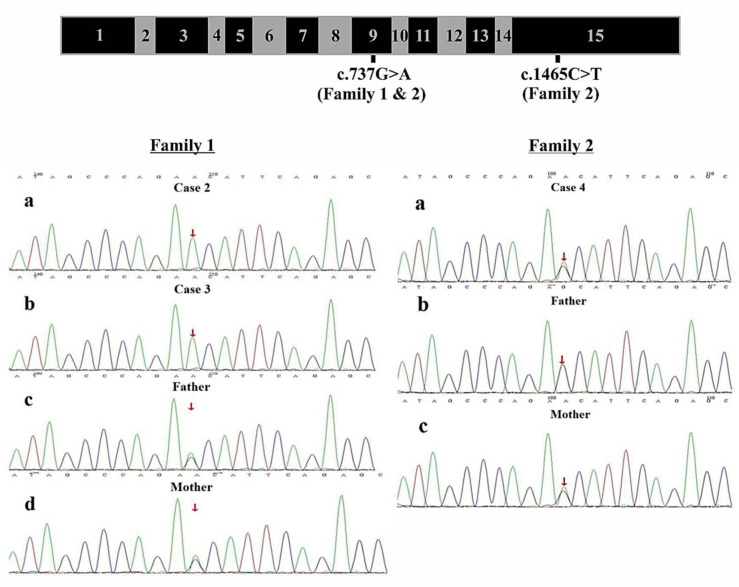
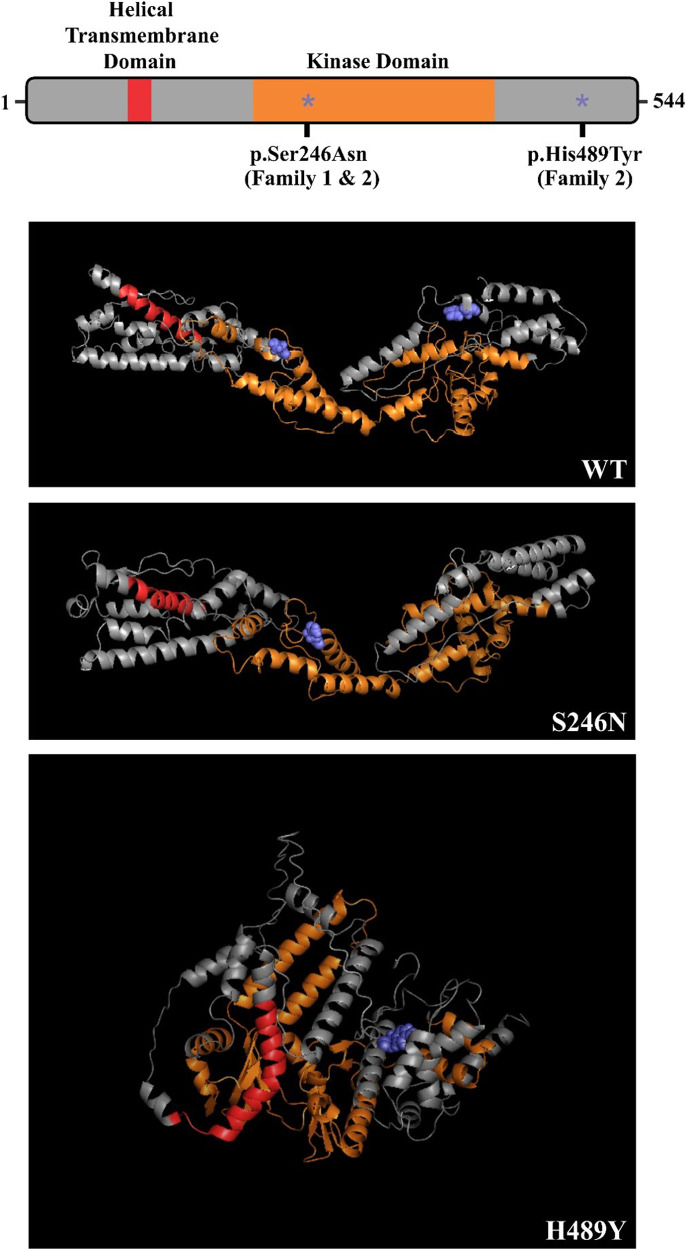
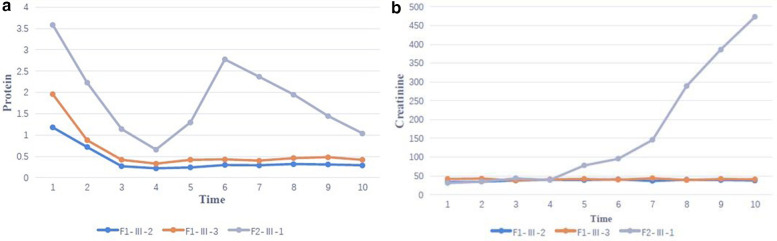
Results: Clinical exome sequencing revealed biallelic variants of the COQ8B gene in 2 families. In the Family 1, the oldest of three affected siblings died of renal failure at 11 years of age. Based on the results of genetic testing which identified a homozygous variant of COQ8B, the other two affected siblings with mild proteinuria and normal renal function were treated with CoQ10 oral supplementation at an early stage. Coenzyme Q10 treatment was effective in reducing proteinuria levels in both patients from Family 1 over the first 6 months and the two patients still have low-level proteinuria and normal renal function at nearly three years. In Family 2, clinical exome sequencing revealed a compoundheterozygous variants of COQ8B in a patient with biopsy- proven FSGS. His disease was unresponsive to prior treatment with glucocorticoids and cyclosporine. Oral CoQ10 was initiated based on his genetic diagnosis and was it was effective in reducing proteinuria over the first 5 months months of therapy. However after 1 year, his disease progressed tokidney failure. Kidney transplantation was performed at 5 years of age and his condition has been stable without rejection and no recurrence of disease.
Conclusions: COQ8B gene variant-related glomerulopathy often presents as SRNS without obvious extrarenal manifestations. The histopathology is mainly FSGS and follows an autosomal recessive mode of inheritance. Some patients may benefit from early coenzyme Q10 supplementation. For patients whose disease progresses to kidney failure, kidney transplantation can be an effective treatment. For children with unexplained proteinuria and abnormal renal function, genetic testing should be performed early in the course of disease to guide therapy where possible and improve prognosis.
Keywords: COQ8B variant; autosomal recessive; focal segmental glomerulosclerosis; glomerulopathy; steroid-resistant nephrotic syndrome.
© 2025 Zhang, Hall, Han, Li and Cui.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Hemophilia B.2000 Oct 2 [updated 2024 Jun 6]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2000 Oct 2 [updated 2024 Jun 6]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301668 Free Books & Documents. Review.
-
Defining the optimum strategy for identifying adults and children with coeliac disease: systematic review and economic modelling.Health Technol Assess. 2022 Oct;26(44):1-310. doi: 10.3310/ZUCE8371. Health Technol Assess. 2022. PMID: 36321689 Free PMC article.
-
Adenosine Deaminase Deficiency.2006 Oct 3 [updated 2024 Mar 7]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2006 Oct 3 [updated 2024 Mar 7]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301656 Free Books & Documents. Review.
-
Sickle Cell Disease.2003 Sep 15 [updated 2025 Feb 13]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2003 Sep 15 [updated 2025 Feb 13]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301551 Free Books & Documents. Review.
-
FBN1-Related Marfan Syndrome.2001 Apr 18 [updated 2022 Feb 17]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2001 Apr 18 [updated 2022 Feb 17]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301510 Free Books & Documents. Review.
References
Grants and funding
LinkOut - more resources
Full Text Sources