

A case report of classic galactosemia with a GALT gene variant and a literature review
- PMID: 38778342
- PMCID: PMC11110268
- DOI: 10.1186/s12887-024-04769-0
A case report of classic galactosemia with a GALT gene variant and a literature review
Abstract
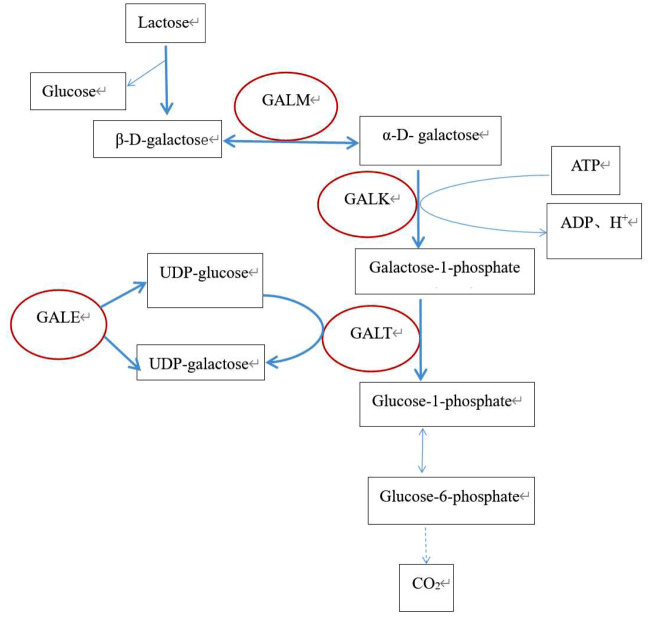
Background: Galactosemia is an autosomal recessive disorder resulting from an enzyme defect in the galactose metabolic pathway. The most severe manifestation of classic galactosemia is caused by galactose-1-phosphate uridylyltransferase (GALT) deficiency, and this condition can be fatal during infancy if left untreated. It also may result in long-term complications in affected individuals.
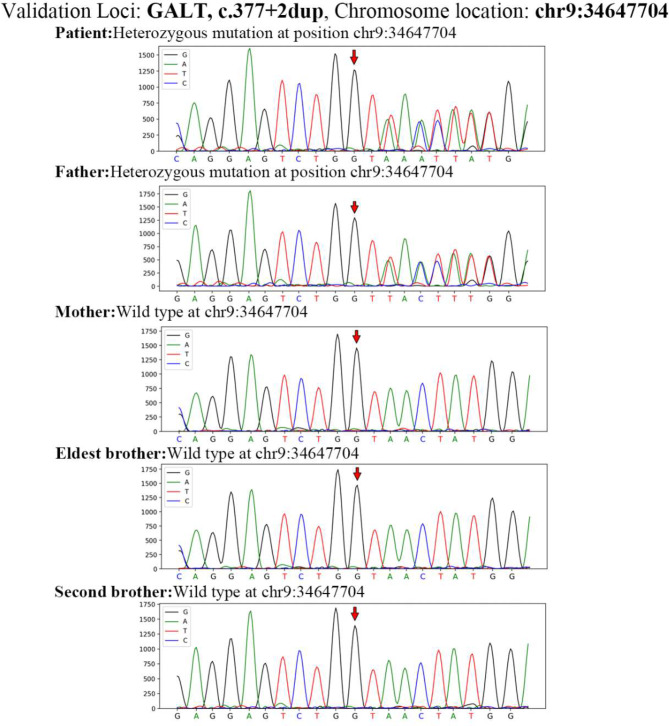
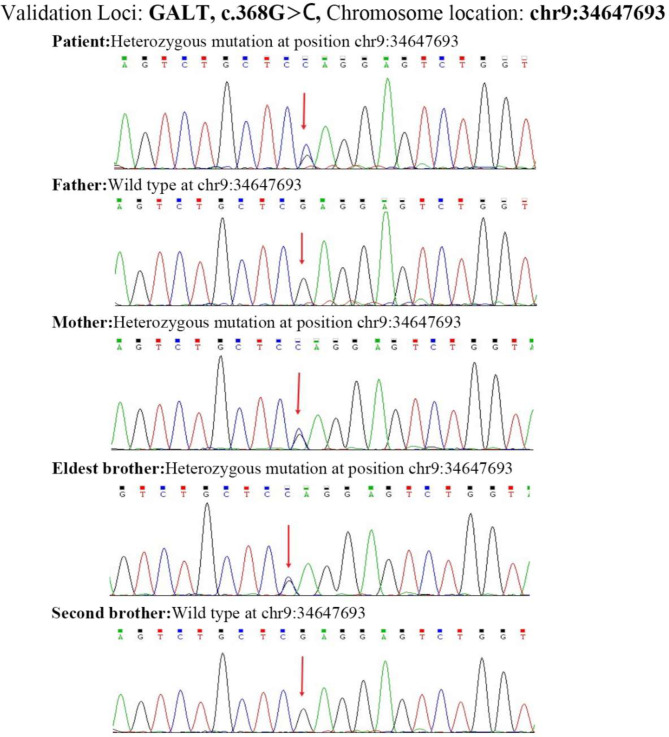
Case presentation: This report describes a patient whose initial clinical symptoms were jaundice and liver dysfunction. The patient's liver and coagulation functions did not improve after multiple admissions and treatment with antibiotics, hepatoprotective and choleretic agents and blood transfusion. Genetic analysis revealed the presence of two variants in the GALT gene in the compound heterozygous state: c.377 + 2dup and c.368G > C (p.Arg123Pro). Currently, the variant locus (c.377 + 2dup) in the GALT gene has not been reported in the Human Gene Mutation Database (HGMD), while c.368G > C (p.Arg123Pro) has not been reported in the Genome Aggregation Database (GnomAD) nor the HGMD in East Asian population. We postulated that the two variants may contribute to the development of classical galactosemia.
Conclusions: Applications of whole-exome sequencing to detect the two variants can improve the detection and early diagnosis of classical galactosemia and, more specifically, may identify individuals who are compound heterozygous with variants in the GALT gene. Variants in the GALT gene have a potential therapeutic significance for classical galactosemia.
Keywords: GALT gene; Exome sequencing; Galactosemia; Literature review; Liver failure.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Galactosemia: when is it a newborn screening emergency?Mol Genet Metab. 2012 May;106(1):7-11. doi: 10.1016/j.ymgme.2012.03.007. Epub 2012 Mar 21. Mol Genet Metab. 2012. PMID: 22483615 Review.
-
Classic Galactosemia: Clinical and Computational Characterization of a Novel GALT Missense Variant (p.A303D) and a Literature Review.Int J Mol Sci. 2023 Dec 12;24(24):17388. doi: 10.3390/ijms242417388. Int J Mol Sci. 2023. PMID: 38139222 Free PMC article. Review.
-
Two Lithuanian Cases of Classical Galactosemia with a Literature Review: A Novel GALT Gene Mutation Identified.Medicina (Kaunas). 2020 Oct 25;56(11):559. doi: 10.3390/medicina56110559. Medicina (Kaunas). 2020. PMID: 33113773 Free PMC article. Review.
-
Novel Mutation in GALT Gene in Galactosemia Patient with Group B Streptococcus Meningitis and Acute Liver Failure.Medicina (Kaunas). 2019 Apr 4;55(4):91. doi: 10.3390/medicina55040091. Medicina (Kaunas). 2019. PMID: 30987402 Free PMC article.
-
Simultaneous occurrence of various mutations and polymorphisms in cis and in trans of the galactose-1-phosphate uridyltransferase gene in a Turkish family with classical galactosemia.J Mol Med (Berl). 1998 Sep;76(10):715-9. doi: 10.1007/s001090050272. J Mol Med (Berl). 1998. PMID: 9766850
References
-
- Cocanougher B, Aypar U, McDonald A, Hasadsri L, Bennett MJ, Highsmith WE, D’Aco K. Compound heterozygosity with a novel S222N GALT mutation leads to atypical galactosemia with loss of GALT activity in erythrocytes but little evidence of clinical disease. Mol Genet Metabolism Rep. 2015;2:61–4. doi: 10.1016/j.ymgmr.2014.12.004. - DOI - PMC - PubMed
-
- Welling L, Bernstein LE, Berry GT, Burlina AB, Eyskens F, Gautschi M, Grünewald S, Gubbels CS, Knerr I, Labrune P, van der Lee JH. International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up. J Inherit Metab Dis. 2017;40:171–6. doi: 10.1007/s10545-016-9990-5. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
