

Indigenous Australian genomes show deep structure and rich novel variation
- PMID: 38093005
- PMCID: PMC10733150
- DOI: 10.1038/s41586-023-06831-w
Indigenous Australian genomes show deep structure and rich novel variation
Abstract
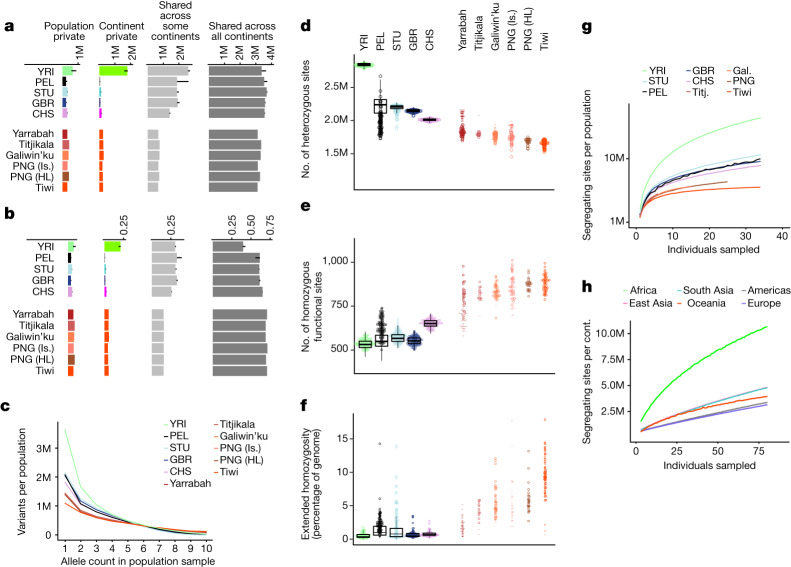
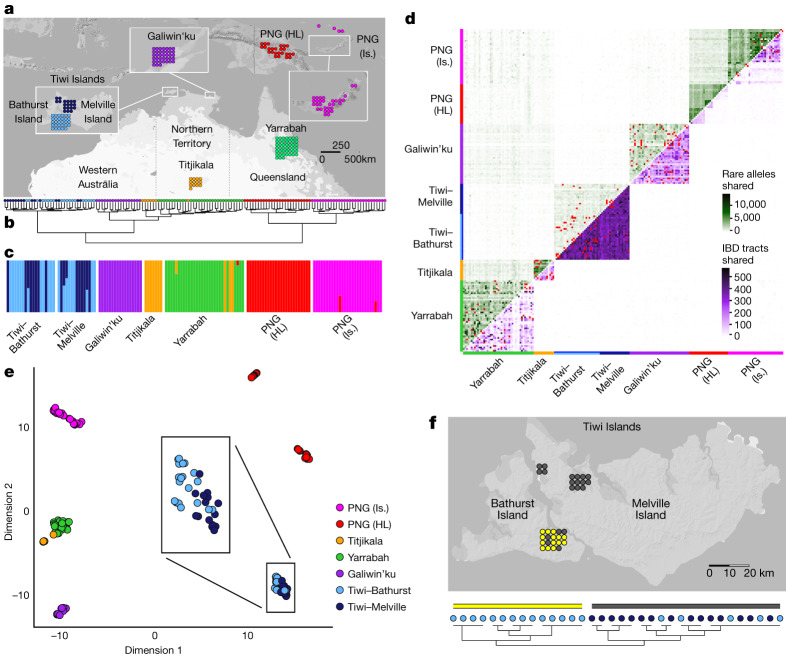
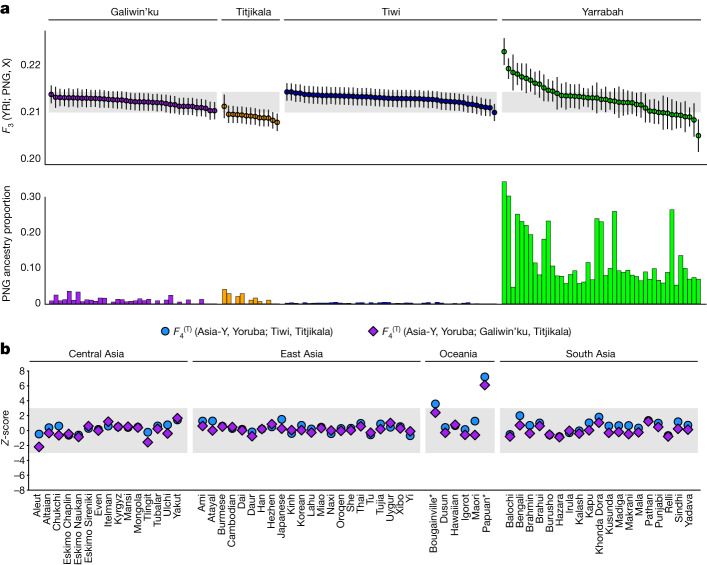
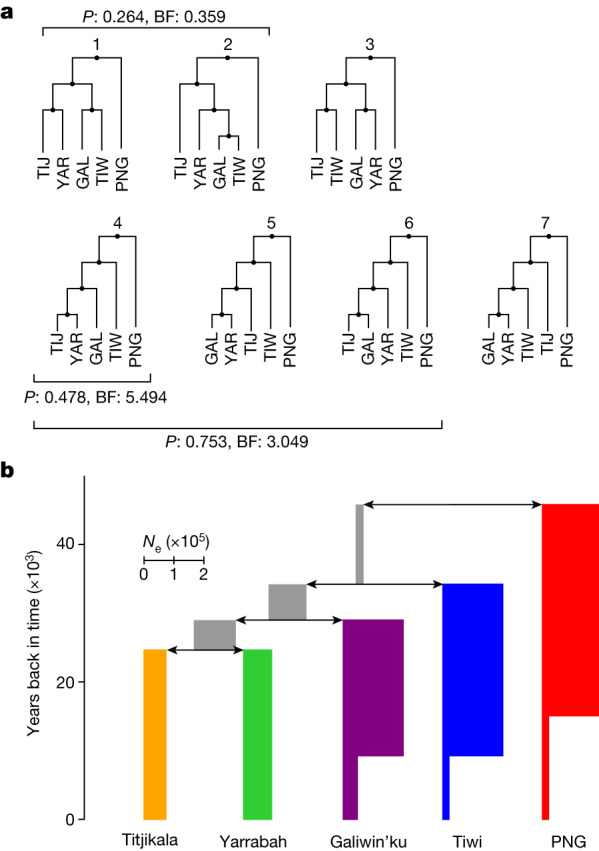
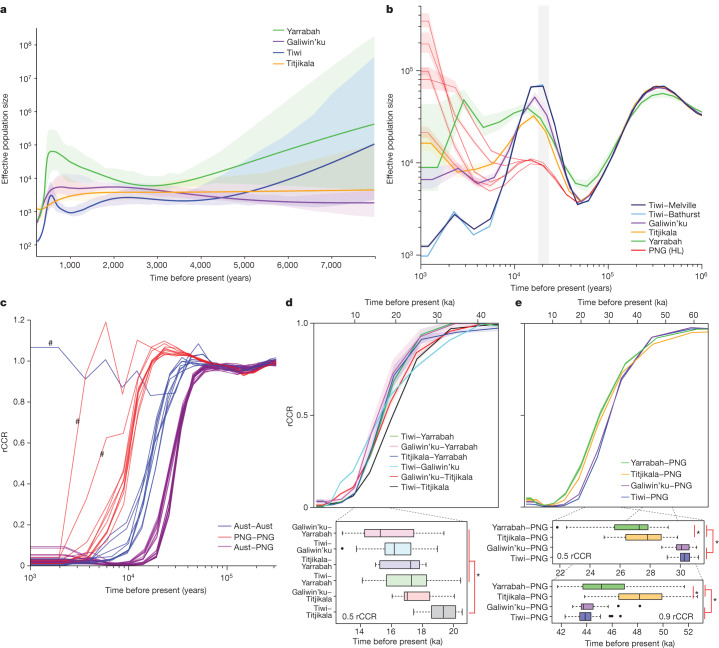
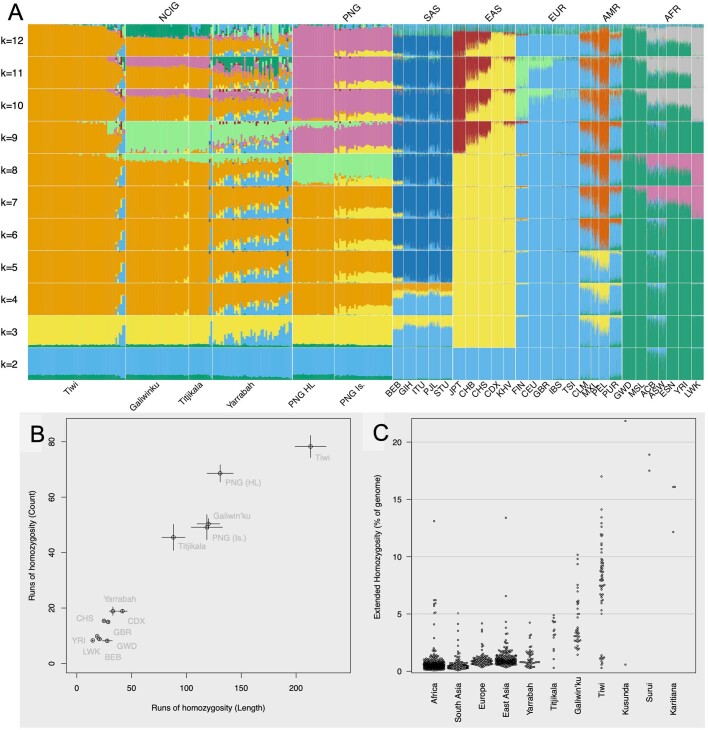
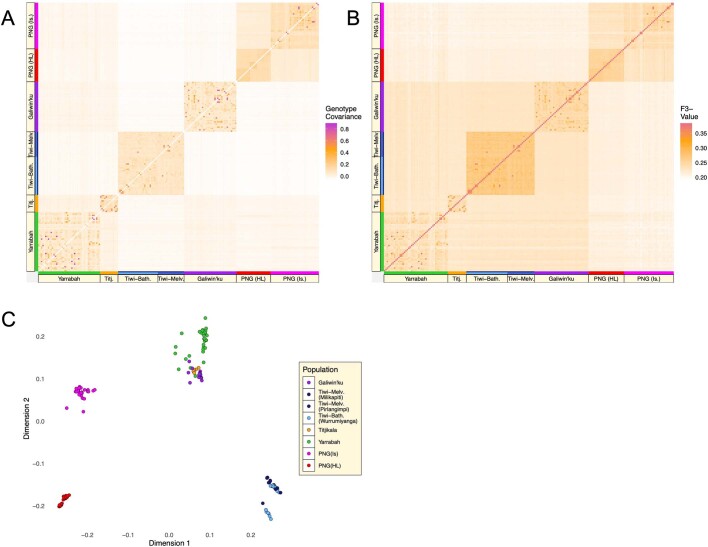
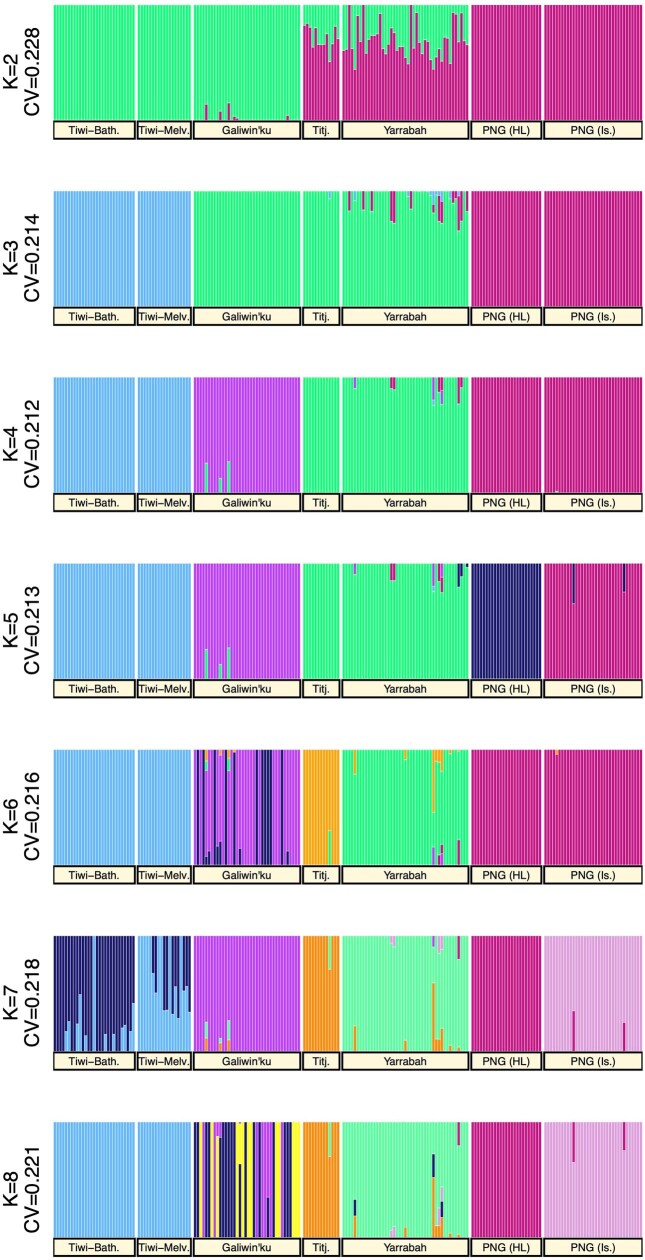
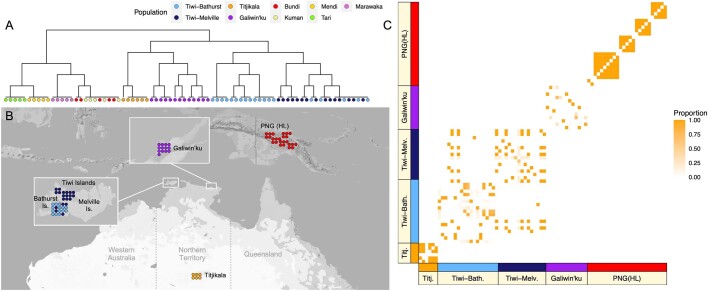
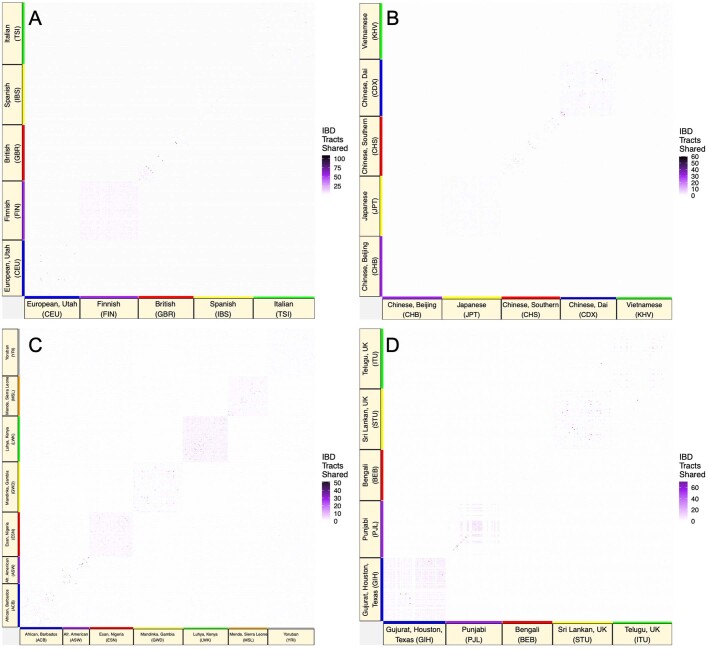
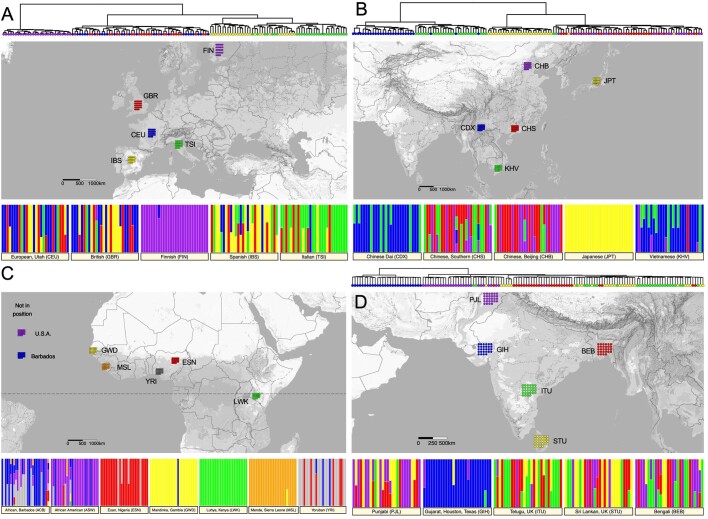
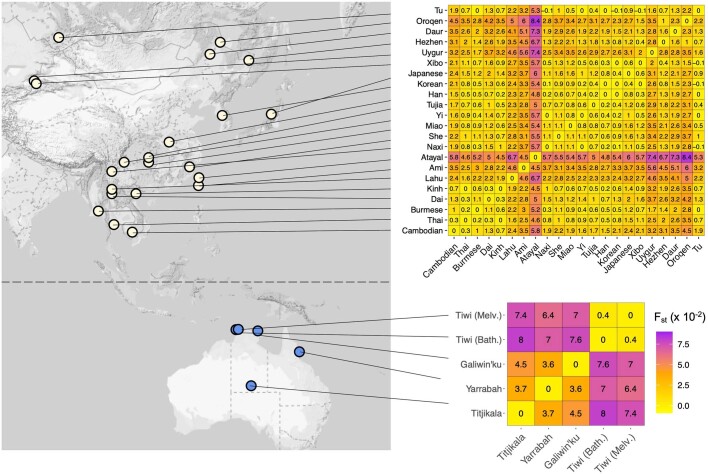
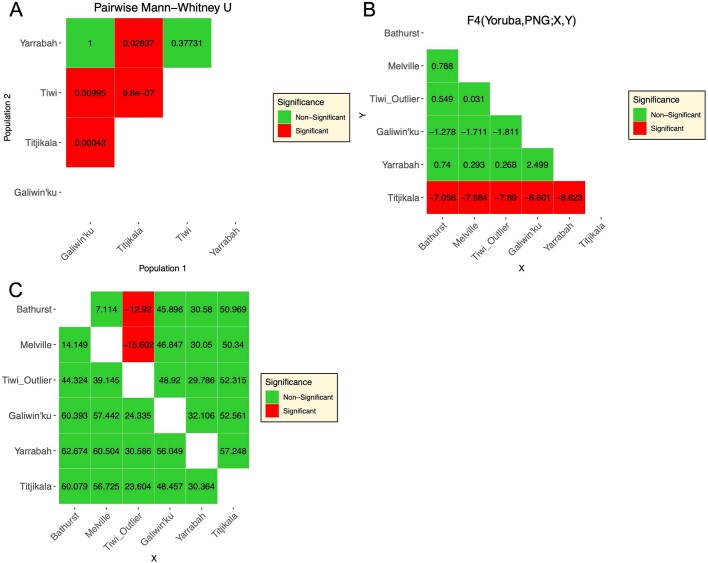
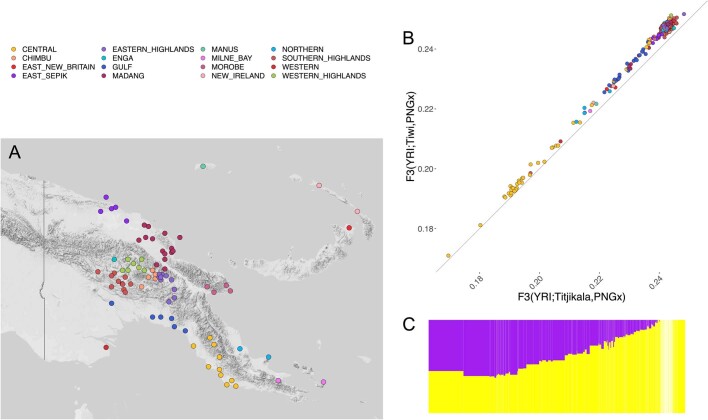
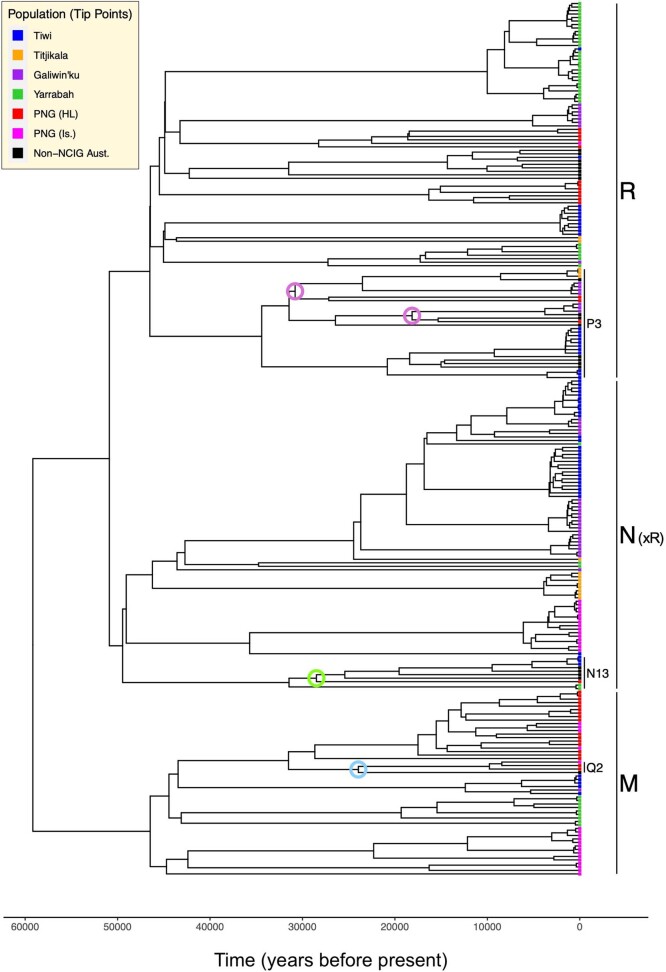
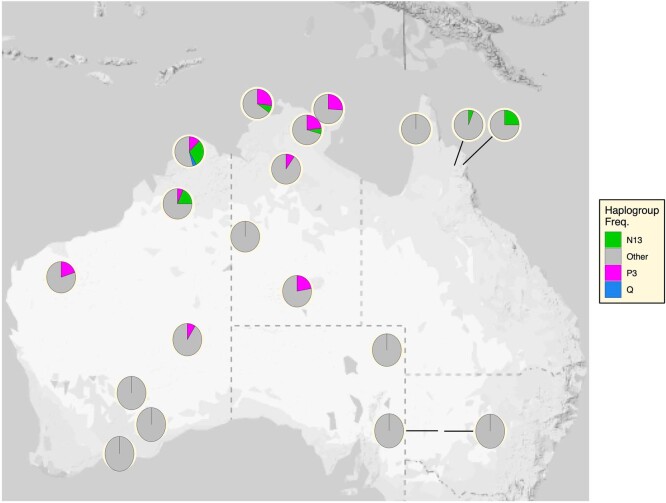
The Indigenous peoples of Australia have a rich linguistic and cultural history. How this relates to genetic diversity remains largely unknown because of their limited engagement with genomic studies. Here we analyse the genomes of 159 individuals from four remote Indigenous communities, including people who speak a language (Tiwi) not from the most widespread family (Pama-Nyungan). This large collection of Indigenous Australian genomes was made possible by careful community engagement and consultation. We observe exceptionally strong population structure across Australia, driven by divergence times between communities of 26,000-35,000 years ago and long-term low but stable effective population sizes. This demographic history, including early divergence from Papua New Guinean (47,000 years ago) and Eurasian groups1, has generated the highest proportion of previously undescribed genetic variation seen outside Africa and the most extended homozygosity compared with global samples. A substantial proportion of this variation is not observed in global reference panels or clinical datasets, and variation with predicted functional consequence is more likely to be homozygous than in other populations, with consequent implications for medical genomics2. Our results show that Indigenous Australians are not a single homogeneous genetic group and their genetic relationship with the peoples of New Guinea is not uniform. These patterns imply that the full breadth of Indigenous Australian genetic diversity remains uncharacterized, potentially limiting genomic medicine and equitable healthcare for Indigenous Australians.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Australian Indigenous genomes are highly diverse and unlike those anywhere else.Nature. 2024 Jan;625(7993):15-16. doi: 10.1038/d41586-023-04006-1. Nature. 2024. PMID: 38093071 No abstract available.
-
The landscape of genomic structural variation in Indigenous Australians.Nature. 2023 Dec;624(7992):602-610. doi: 10.1038/s41586-023-06842-7. Epub 2023 Dec 13. Nature. 2023. PMID: 38093003 Free PMC article.
-
Novel genetic markers for chronic kidney disease in a geographically isolated population of Indigenous Australians: Individual and multiple phenotype genome-wide association study.Genome Med. 2024 Feb 12;16(1):29. doi: 10.1186/s13073-024-01299-3. Genome Med. 2024. PMID: 38347632 Free PMC article.
-
From biocolonialism to emancipation: considerations on ethical and culturally respectful omics research with indigenous Australians.Med Health Care Philos. 2023 Sep;26(3):487-496. doi: 10.1007/s11019-023-10151-1. Epub 2023 May 12. Med Health Care Philos. 2023. PMID: 37171744 Free PMC article. Review.
-
The prevelance of psychosis in indigenous populations in Australia: A review of the literature using systematic methods.Australas Psychiatry. 2023 Jun;31(3):376-380. doi: 10.1177/10398562231156317. Epub 2023 Feb 8. Australas Psychiatry. 2023. PMID: 36753669 Review.
Cited by
-
Precision medicine in Australia: indigenous health professionals are needed to improve equity for Aboriginal and Torres Strait Islanders.Int J Equity Health. 2024 Jul 4;23(1):134. doi: 10.1186/s12939-024-02202-7. Int J Equity Health. 2024. PMID: 38965527 Free PMC article.
-
Australian Indigenous genomes are highly diverse and unlike those anywhere else.Nature. 2024 Jan;625(7993):15-16. doi: 10.1038/d41586-023-04006-1. Nature. 2024. PMID: 38093071 No abstract available.
-
Increasing Diversity, Equity, Inclusion, and Accessibility in Rare Disease Clinical Trials.Pharmaceut Med. 2024 Jul;38(4):261-276. doi: 10.1007/s40290-024-00529-8. Epub 2024 Jul 9. Pharmaceut Med. 2024. PMID: 38977611 Review.
-
A call to action to scale up research and clinical genomic data sharing.Nat Rev Genet. 2025 Feb;26(2):141-147. doi: 10.1038/s41576-024-00776-0. Epub 2024 Oct 7. Nat Rev Genet. 2025. PMID: 39375561 Review.
-
Estimating evolutionary and demographic parameters via ARG-derived IBD.bioRxiv [Preprint]. 2024 Mar 13:2024.03.07.583855. doi: 10.1101/2024.03.07.583855. bioRxiv. 2024. Update in: PLoS Genet. 2025 Jan 8;21(1):e1011537. doi: 10.1371/journal.pgen.1011537. PMID: 38559261 Free PMC article. Updated. Preprint.
References
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
