

[Recommendations for the healthcare of patients with FOP]
- PMID: 37603129
- PMCID: PMC10622346
- DOI: 10.1007/s00132-023-04425-y
[Recommendations for the healthcare of patients with FOP]
Abstract
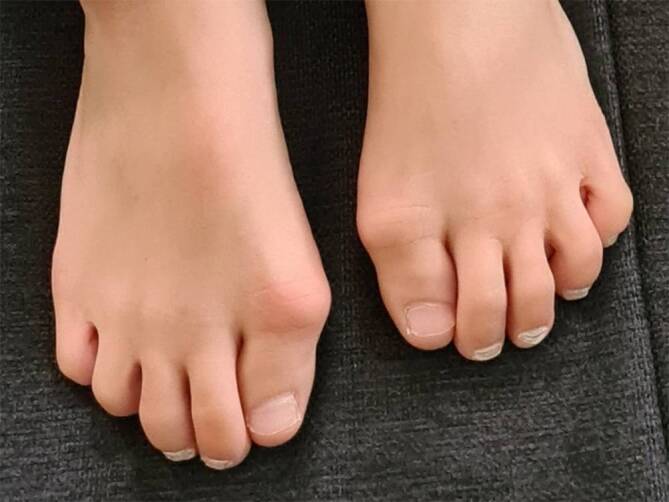
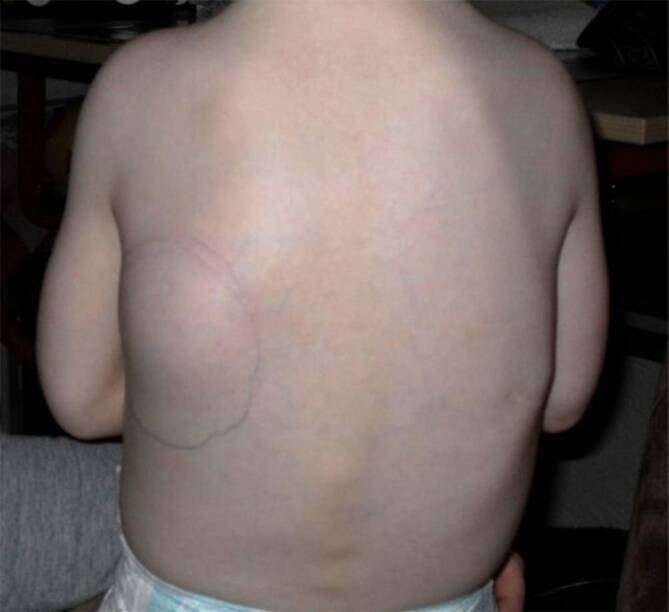
Background: Fibrodysplasia ossificans progressiva (FOP) is a very rare, severe genetic disorder triggered by a gain-of-function mutation in the ACVR1 gene that codes for the type I bone morphogenetic protein (BMP) receptor ACVR1 (activin A receptor-type 1), also known as ALK2 (activin receptor-like kinase-2). It leads to the onset and progression of heterotopic ossification (HO) in soft and connective tissue. HO is often preceded by episodes of soft tissue swelling or flare-ups. Flare-ups, characteristic of FOP, may be induced by trauma, infection, vaccination, or other medications, as well as surgical procedures or may occur spontaneously. As patients age, they develop severe mobility limitations due to progressive HO formation, including immobility, causing a shortened life expectancy. FOP's first characteristic clinical sign is the congenital malformation of one or both big toes with valgus axis deviation, which is present in almost all patients. To confirm the diagnosis, molecular genetic analysis of the ACVR1 gene is possible.
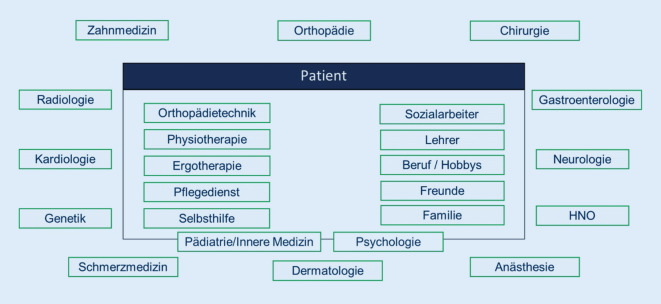
Aim of the recommendations: This white paper aims to provide an overview of the necessary prerequisites and conditions for the care of patients with FOP and positively contribute to patients with FOP by improving the overall availability of knowledge. To achieve this, relevant aspects of the care of the very rare disease FOP are presented, from the initial diagnosis to the care in regular care based on the authors' knowledge (German FOP network) and the international FOP Treatment Guidelines. The recommendations presented here are addressed to all actors and decision-makers in the health care system and are also intended to inform patients and the public.
Zusammenfassung: HINTERGRUND: Bei der Fibrodysplasia ossificans progressiva (FOP) handelt es sich um eine sehr seltene, genetisch bedingte Erkrankung, ausgelöst durch eine „Gain-of-function“-Mutation im ACVR1-Gen, welches den Typ-I-Bone-Morphogenetic-Protein(BMP)-Rezeptor ACVR1 („activin A receptor type 1“) – auch bekannt als ALK2 („activin receptor-like kinase 2“) kodiert. Diese Mutation führt zum Auftreten und Fortschreiten heterotoper Ossifikationen (HO) im Weich- und Bindegewebe. Der HO gehen oft Episoden von Weichteilschwellungen, sogenannte Flare-ups voraus. Die für FOP charakteristischen Flare-ups können durch Traumata, Infektionen, Impfungen oder andere medizinische sowie chirurgische Eingriffe induziert werden oder spontan auftreten. Mit fortschreitendem Alter der Patient:innen kommt es bei den Betroffenen aufgrund zunehmender HO zu schwerwiegenden Bewegungseinschränkungen bis hin zur Bewegungsunfähigkeit, die mit einer verkürzten Lebenserwartung einhergeht. Ein erstes charakteristisches klinisches Anzeichen für FOP ist die angeborene Fehlbildung der Großzehen [25] mit valgischer Achsabweichung, die bei fast allen Patient:innen auftritt. Um die Diagnose zu sichern, ist eine molekulargenetische Analyse des ACVR1-Gens möglich.
Ziel der empfehlungen: Ziel der vorliegenden Handlungsempfehlungen ist es, einen Überblick über die notwendigen Voraussetzungen und Bedingungen für die Versorgung von Patient:innen mit FOP zu geben und durch eine bessere Verfügbarkeit von Wissen insgesamt einen positiven Beitrag für Patient:innen mit FOP zu leisten. Um dies zu erreichen, werden relevante Aspekte bei der Versorgung der sehr seltenen Erkrankung FOP vorgestellt, von der initialen Diagnose bis zur Betreuung in der Regelversorgung, basierend auf dem Wissen der Autor:innen (deutsches FOP-Netzwerk) und den internationalen FOP Treatment Guidelines. Die hier vorgestellten Empfehlungen richten sich an alle Akteur:innen und Entscheidungsträger:innen im Gesundheitswesen und sollen darüber hinaus der Information von Betroffenen und der Öffentlichkeit dienen.
Keywords: Fibrodysplasia ossificans progressiva; Heterotopic ossifications; Myositis ossificans progressiva; Rare disease; Symptom flare-up.
© 2023. The Author(s).
Similar articles
-
The serum levels of activin A and bone morphogenetic protein-4 and -6 in patients with fibrodysplasia ossificans progressiva.Orphanet J Rare Dis. 2023 May 10;18(1):111. doi: 10.1186/s13023-023-02708-3. Orphanet J Rare Dis. 2023. PMID: 37165433 Free PMC article.
-
Depletion of Mast Cells and Macrophages Impairs Heterotopic Ossification in an Acvr1R206H Mouse Model of Fibrodysplasia Ossificans Progressiva.J Bone Miner Res. 2018 Feb;33(2):269-282. doi: 10.1002/jbmr.3304. Epub 2018 Jan 3. J Bone Miner Res. 2018. PMID: 28986986 Free PMC article.
-
Fibrodysplasia ossificans progressiva: clinical and genetic aspects.Orphanet J Rare Dis. 2011 Dec 1;6:80. doi: 10.1186/1750-1172-6-80. Orphanet J Rare Dis. 2011. PMID: 22133093 Free PMC article. Review.
-
Fibrodysplasia Ossificans Progressiva: A Challenging Diagnosis.Genes (Basel). 2021 Jul 30;12(8):1187. doi: 10.3390/genes12081187. Genes (Basel). 2021. PMID: 34440363 Free PMC article.
-
BMP signaling and skeletal development in fibrodysplasia ossificans progressiva (FOP).Dev Dyn. 2022 Jan;251(1):164-177. doi: 10.1002/dvdy.387. Epub 2021 Jun 26. Dev Dyn. 2022. PMID: 34133058 Free PMC article. Review.
References
-
- https://clinicaltrials.gov/ct2/results?cond=fibrodysplasia+ossificans+pr.... Zugegriffen: 31. März 2023
-
- https://www.ifopa.org/2020_fop_registry_annual_report. Zugegriffen: 19. März 2023
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials