

Disease Modifying Therapies for the Management of Children with Spinal Muscular Atrophy (5q SMA): An Update on the Emerging Evidence
- PMID: 35734367
- PMCID: PMC9208376
- DOI: 10.2147/DDDT.S214174
Disease Modifying Therapies for the Management of Children with Spinal Muscular Atrophy (5q SMA): An Update on the Emerging Evidence
Abstract
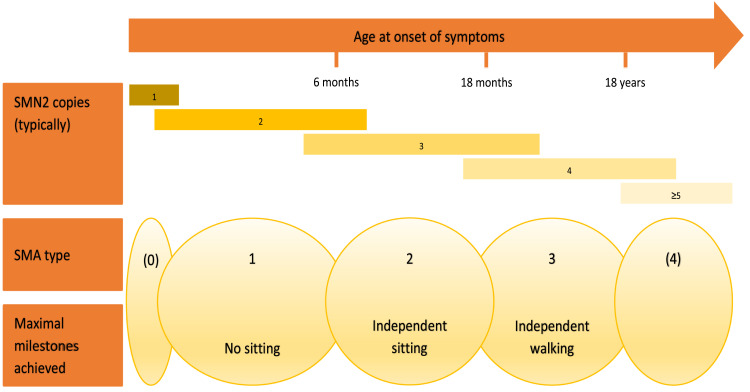
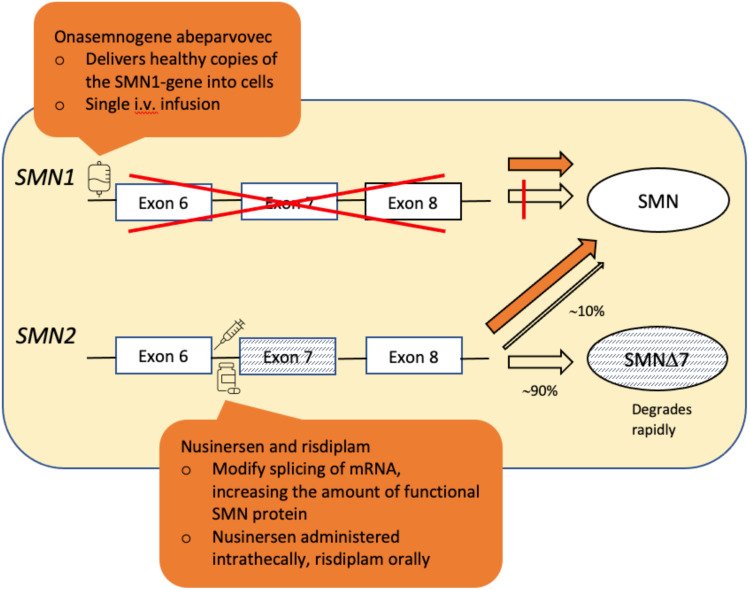
SMA (5q SMA) is an autosomal recessive neuromuscular disease with an estimated incidence of approximately 1 in 11,000 live births, characterized by progressive degeneration and loss of α-motor neurons in the spinal cord and brain stem, resulting in progressive muscle weakness. The disease spectrum is wide, from a serious congenital to a mild adult-onset disease. SMA is caused by biallelic mutations in the SMN1 gene and disease severity is modified primarily by SMN2 copy number. Before the advent of specific disease altering treatments, SMA was the second most common fatal autosomal recessive disorder after cystic fibrosis and the most common genetic cause of infant mortality. Nusinersen, risdiplam, and onasemnogene abeparvovec are presently the only approved disease modifying therapies for SMA, and the aim of this review is to discuss their mode of action, effects, safety concerns, and results from real-world experience. All exert their action by increasing the level of SMN protein in lower motor neuron. Nusinersen and risdiplam by modifying the SMN2 gene product, and onasemnogene abeparvovec by delivering SMN1 gene copies into cells. All have an established clinical efficacy. An important feature shared by all three is that early intervention is associated with a better treatment outcome, such that in cases where treatment is initiated in an early pre-symptomatic period, it may result in normal - or almost normal - motor development. Thus, early diagnosis followed by swift initiation of treatment is fundamental for the treatment response and consequently long-term prognosis in SMA type 1, and probably SMA type 2. The same principle similarly applies to the milder phenotypes. All three therapies are relatively novel, with risdiplam being the latest addition. Except for nusinersen, real-world data are still scarce, and long-term data are quite naturally lacking.
Keywords: disease-modifying; gene therapy; spinal muscular atrophy; treatment.
© 2022 Hjartarson et al.
Conflict of interest statement
Thomas Sejersen: Recipient of honoraria received for lectures or consultancy from Biogen, Novartis, PTC Therapeutics, Sarepta Therapeutics, Roche, Hansa Biopharma and Sanofi Genzyme. The authors report no other conflicts of interest in this work.
Figures
Similar articles
-
Nusinersen: A Review in 5q Spinal Muscular Atrophy.CNS Drugs. 2021 Dec;35(12):1317-1328. doi: 10.1007/s40263-021-00878-x. Epub 2021 Nov 30. CNS Drugs. 2021. PMID: 34850360 Free PMC article.
-
Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy.Cells. 2022 Jan 26;11(3):417. doi: 10.3390/cells11030417. Cells. 2022. PMID: 35159227 Free PMC article. Review.
-
[Risdiplam for the treatment of spinal muscular atrophy].Zh Nevrol Psikhiatr Im S S Korsakova. 2024;124(2):45-57. doi: 10.17116/jnevro202412402145. Zh Nevrol Psikhiatr Im S S Korsakova. 2024. PMID: 38465810 Russian.
-
Health Care Resource Utilization and Costs for Patients with Spinal Muscular Atrophy: Findings from a Retrospective US Claims Database Analysis.Adv Ther. 2023 Oct;40(10):4589-4605. doi: 10.1007/s12325-023-02621-y. Epub 2023 Aug 16. Adv Ther. 2023. PMID: 37587305 Free PMC article.
-
Recent Advance in Disease Modifying Therapies for Spinal Muscular Atrophy.Acta Neurol Taiwan. 2024 Sep 30;33(3):81-88. Acta Neurol Taiwan. 2024. PMID: 39363429 Review.
Cited by
-
A Mixed-method Approach to Develop an Ambulatory Module of the SMA Independence Scale.J Neuromuscul Dis. 2023;10(6):1093-1109. doi: 10.3233/JND-230096. J Neuromuscul Dis. 2023. PMID: 37742658 Free PMC article.
-
Sex Difference in Spinal Muscular Atrophy Patients - are Males More Vulnerable?J Neuromuscul Dis. 2023;10(5):847-867. doi: 10.3233/JND-230011. J Neuromuscul Dis. 2023. PMID: 37393514 Free PMC article.
-
No significant sex differences in incidence or phenotype for the SMNΔ7 mouse model of spinal muscular atrophy.Neuromuscul Disord. 2024 Apr;37:13-22. doi: 10.1016/j.nmd.2024.03.002. Epub 2024 Mar 5. Neuromuscul Disord. 2024. PMID: 38493520
-
Nusinersen Improves Motor Function in Type 2 and 3 Spinal Muscular Atrophy Patients across Time.Biomedicines. 2024 Aug 6;12(8):1782. doi: 10.3390/biomedicines12081782. Biomedicines. 2024. PMID: 39200246 Free PMC article.
-
Healthcare resource utilisation and direct medical cost for individuals with 5q spinal muscular atrophy in Sweden.Eur J Health Econ. 2025 Feb;26(1):35-48. doi: 10.1007/s10198-024-01678-y. Epub 2024 Apr 20. Eur J Health Econ. 2025. PMID: 38642267 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
