

Primary hyperoxaluria type 1: novel therapies at a glance
- PMID: 35592618
- PMCID: PMC9113449
- DOI: 10.1093/ckj/sfab245
Primary hyperoxaluria type 1: novel therapies at a glance
Abstract
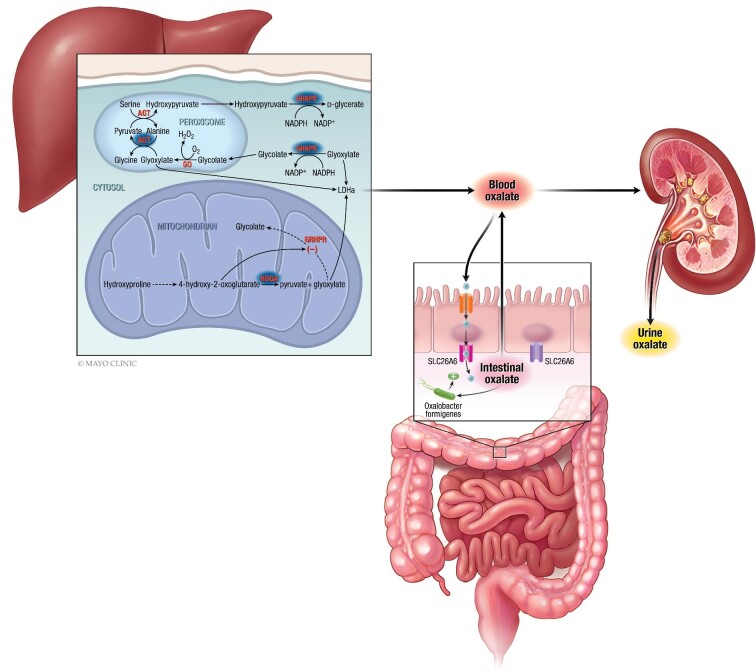
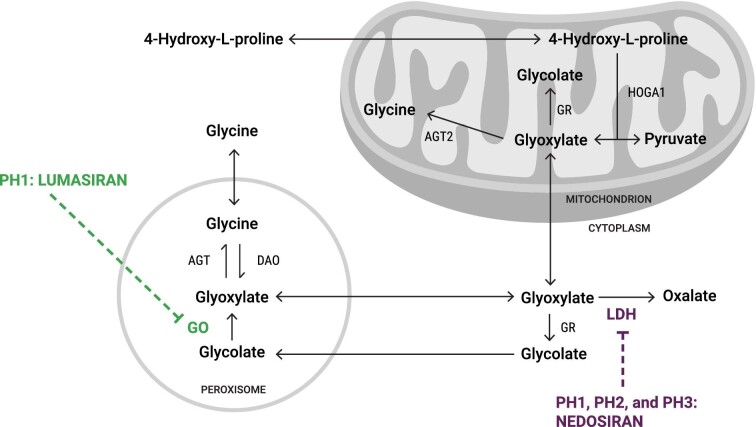
Primary hyperoxaluria type 1 (PH1) is a rare and severe autosomal recessive disease of oxalate metabolism, resulting from a mutation in the AGXT gene that encodes the hepatic peroxisomal enzyme alanine-glyoxylate aminotransferase (AGT). Until recently, treatment of PH1 was supportive, consisting of intensive hyperhydration, use of crystallization inhibitors (citrate and neutral phosphorus), in a subset of responsive PH1 patients' pharmacologic doses of vitamin B6 (pyridoxine), and kidney and liver transplantation when patients progressed to kidney failure. Treatment approaches have been similar for PH2 caused by mutations in hepatic glyoxylate reductase/hydroxypyruvate reductase (GR/HPR), although pyridoxine does not have any benefit in this group. PH3 is caused by mutations of mitochondrial 4-hydroxy-2-oxoglutarate aldolase (HOGA1) and was the most recently described. Kidney failure appears less common in PH3, although kidney stones occur as frequently as in PH1 and PH2. Oxalate metabolism in the liver is complex. Novel therapies based on RNA interference (RNAi) have recently emerged to modulate these pathways, designed to deplete substrate for enzymes upstream and decrease/avoid oxalate production. Two hepatic enzymes have been targeted to date in PH: glycolate oxidase (GO) with lumasiran and lactate dehydrogenase A (LDH-A) with nedosiran. Lumasiran was approved for the treatment of PH1 in 2020 by both the European Medicines Agency and the Food and Drug Administration, whilst clinical trials with nedosiran are ongoing. Results with the two RNAi therapies demonstrate a significant reduction of urinary oxalate excretion in PH1 patients, but long-term data on efficacy (preservation of kidney function, decreased stone events) and safety remain to be established. Nevertheless, the hepatically targeted RNAi approach represents a potential 'game changer' in the field of PH1, bringing hope to families and patients that they may be able to avoid liver and/or kidney transplantation in the future and suffer fewer stone events, perhaps with less strict therapeutic regimens. Pharmacological compounds directly inhibiting GO or LDH are also under development and could be of special interest in developing countries where RNAi therapies may not be readily available in the near future. Approaches to manipulate the intestinal microbiome with a goal to increase oxalate degradation or to stimulate secretion of oxalate into the intestine from plasma are also under development. Overall, we appear to be entering a new phase of PH treatment, with an array of promising approaches emerging that will need optimization and evaluation to establish long-term efficacy and safety.
Keywords: LDH-A; RNA interference; glycolate oxidase; hyperoxaluria; paediatrics.
© The Author(s) 2022. Published by Oxford University Press on behalf of the ERA.
Figures
Similar articles
-
Lumasiran for primary hyperoxaluria type 1: What we have learned?Front Pediatr. 2023 Jan 10;10:1052625. doi: 10.3389/fped.2022.1052625. eCollection 2022. Front Pediatr. 2023. PMID: 36704142 Free PMC article. Review.
-
Treatment of primary hyperoxaluria type 1.Clin Kidney J. 2022 May 17;15(Suppl 1):i9-i13. doi: 10.1093/ckj/sfab232. eCollection 2022 May. Clin Kidney J. 2022. PMID: 35592620 Free PMC article. Review.
-
Lumasiran for Advanced Primary Hyperoxaluria Type 1: Phase 3 ILLUMINATE-C Trial.Am J Kidney Dis. 2023 Feb;81(2):145-155.e1. doi: 10.1053/j.ajkd.2022.05.012. Epub 2022 Jul 14. Am J Kidney Dis. 2023. PMID: 35843439 Clinical Trial.
-
New therapeutics for primary hyperoxaluria type 1.Curr Opin Nephrol Hypertens. 2022 Jul 1;31(4):344-350. doi: 10.1097/MNH.0000000000000790. Epub 2022 Mar 9. Curr Opin Nephrol Hypertens. 2022. PMID: 35266883 Free PMC article. Review.
-
Lumasiran: A Review in Primary Hyperoxaluria Type 1.Drugs. 2024 Feb;84(2):219-226. doi: 10.1007/s40265-023-01987-1. Epub 2024 Jan 22. Drugs. 2024. PMID: 38252335 Free PMC article. Review.
Cited by
-
Synthesis of Ethyl Pyrimidine-Quinolincarboxylates Selected from Virtual Screening as Enhanced Lactate Dehydrogenase (LDH) Inhibitors.Int J Mol Sci. 2024 Sep 9;25(17):9744. doi: 10.3390/ijms25179744. Int J Mol Sci. 2024. PMID: 39273691 Free PMC article.
-
Between hope and reality: treatment of genetic diseases through nucleic acid-based drugs.Commun Biol. 2024 Apr 23;7(1):489. doi: 10.1038/s42003-024-06121-9. Commun Biol. 2024. PMID: 38653753 Free PMC article. Review.
-
Nedosiran: First Approval.Drugs. 2023 Dec;83(18):1729-1733. doi: 10.1007/s40265-023-01976-4. Drugs. 2023. PMID: 38060091 Free PMC article. Review.
-
Simultaneous or sequential kidney-liver transplantation in primary hyperoxaluria.J Nephrol. 2024 Oct 9. doi: 10.1007/s40620-024-02109-0. Online ahead of print. J Nephrol. 2024. PMID: 39382784
-
Hydration and Nephrolithiasis in Pediatric Populations: Specificities and Current Recommendations.Nutrients. 2023 Feb 1;15(3):728. doi: 10.3390/nu15030728. Nutrients. 2023. PMID: 36771434 Free PMC article. Review.
References
-
- Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med 2013; 369: 649–658 - PubMed
-
- Harambat J, Fargue S, Acquaviva Cet al. . Genotype–phenotype correlation in primary hyperoxaluria type 1: the p.Gly170Arg AGXT mutation is associated with a better outcome. Kidney Int 2010; 77: 443–449 - PubMed
-
- Fargue S, Harambat J, Gagnadoux M-Fet al. . Effect of conservative treatment on the renal outcome of children with primary hyperoxaluria type 1. Kidney Int 2009; 76: 767–773 - PubMed
-
- Cochat P, Hulton S-A, Acquaviva Cet al. . Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 2012; 27: 1729–1736 - PubMed
-
- Fire A, Xu S, Montgomery MKet al. . Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998; 391: 806–811 - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
