

Newer variants of progressive familial intrahepatic cholestasis
- PMID: 35070006
- PMCID: PMC8727216
- DOI: 10.4254/wjh.v13.i12.2024
Newer variants of progressive familial intrahepatic cholestasis
Abstract
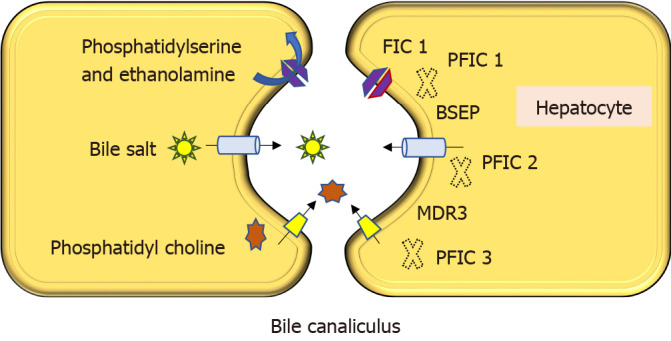
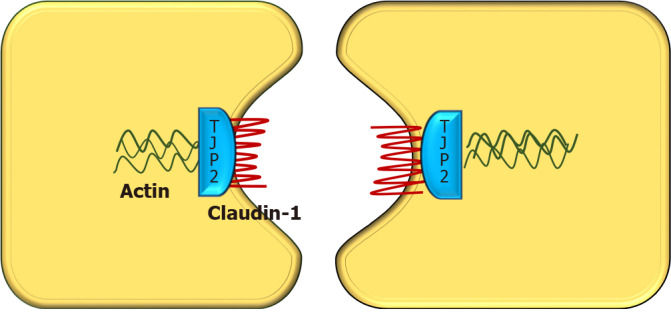
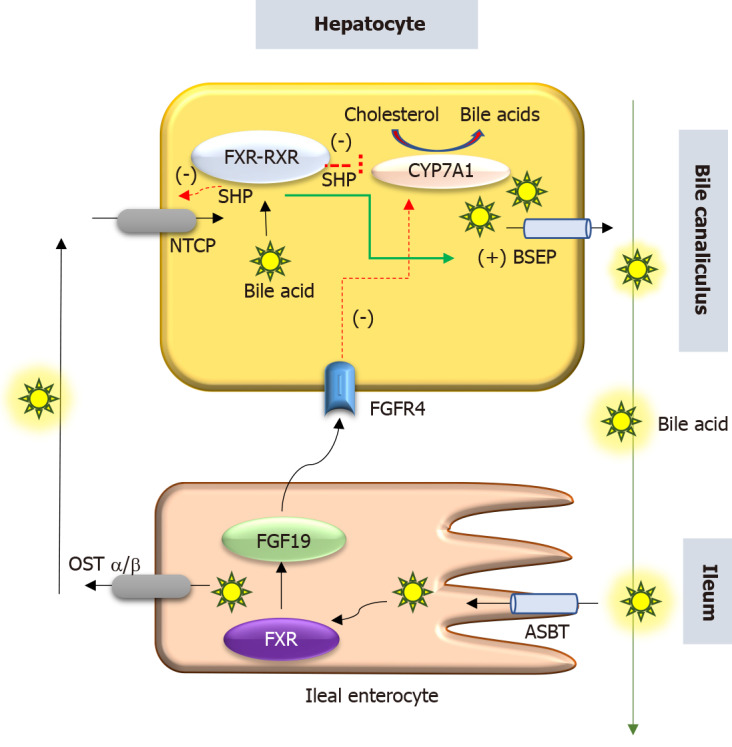
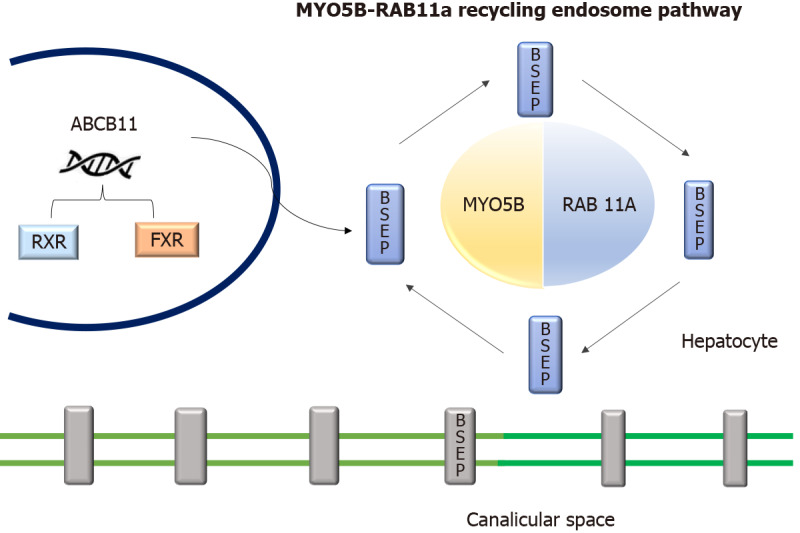
Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of disorders characterized by defects in bile secretion and presentation with intrahepatic cholestasis in infancy or childhood. The most common types include PFIC 1 (deficiency of FIC1 protein, ATP8B1 gene mutation), PFIC 2 (bile salt export pump deficiency, ABCB11 gene mutation), and PFIC 3 (multidrug resistance protein-3 deficiency, ABCB4 gene mutation). Mutational analysis of subjects with normal gamma-glutamyl transferase cholestasis of unknown etiology has led to the identification of newer variants of PFIC, known as PFIC 4, 5, and MYO5B related (sometimes known as PFIC 6). PFIC 4 is caused by the loss of function of tight junction protein 2 (TJP2) and PFIC 5 is due to NR1H4 mutation causing Farnesoid X receptor deficiency. MYO5B gene mutation causes microvillous inclusion disease (MVID) and is also associated with isolated cholestasis. Children with TJP2 related cholestasis (PFIC-4) have a variable spectrum of presentation. Some have a self-limiting disease, while others have progressive liver disease with an increased risk of hepatocellular carcinoma. Hence, frequent surveillance for hepatocellular carcinoma is recommended from infancy. PFIC-5 patients usually have rapidly progressive liver disease with early onset coagulopathy, high alpha-fetoprotein and ultimately require a liver transplant. Subjects with MYO5 B-related disease can present with isolated cholestasis or cholestasis with intractable diarrhea (MVID). These children are at risk of worsening cholestasis post intestinal transplant (IT) for MVID, hence combined intestinal and liver transplant or IT with biliary diversion is preferred. Immunohistochemistry can differentiate most of the variants of PFIC but confirmation requires genetic analysis.
Keywords: Biliary diversion; Hepatocellular carcinoma; Microvillous inclusion disease; Progressive familial intrahepatic cholestasis; Tight junction protein.
©The Author(s) 2021. Published by Baishideng Publishing Group Inc. All rights reserved.
Conflict of interest statement
Conflict-of-interest statement: The authors declare that they do not have any conflict of interest to disclose.
Figures
Similar articles
-
Progressive Familial Intrahepatic Cholestasis: A Study in Children From a Liver Transplant Center in India.J Clin Exp Hepatol. 2022 Mar-Apr;12(2):454-460. doi: 10.1016/j.jceh.2021.06.006. Epub 2021 Jun 13. J Clin Exp Hepatol. 2022. PMID: 35535061 Free PMC article.
-
Molecular overview of progressive familial intrahepatic cholestasis.World J Gastroenterol. 2020 Dec 21;26(47):7470-7484. doi: 10.3748/wjg.v26.i47.7470. World J Gastroenterol. 2020. PMID: 33384548 Free PMC article. Review.
-
Progressive familial intrahepatic cholestasis.J Clin Exp Hepatol. 2014 Mar;4(1):25-36. doi: 10.1016/j.jceh.2013.10.005. Epub 2013 Nov 23. J Clin Exp Hepatol. 2014. PMID: 25755532 Free PMC article. Review.
-
MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease.Hepatology. 2014 Jul;60(1):301-10. doi: 10.1002/hep.26974. Epub 2014 May 27. Hepatology. 2014. PMID: 24375397
-
Clinical outcomes of surgical management for rare types of progressive familial intrahepatic cholestasis: a case series.Surg Case Rep. 2022 Jan 13;8(1):10. doi: 10.1186/s40792-022-01365-1. Surg Case Rep. 2022. PMID: 35024979 Free PMC article.
Cited by
-
Cholestatic Liver Disease due to Novel USP53 Mutations: A Case Series of Three Indian Children.J Clin Exp Hepatol. 2024 Mar-Apr;14(2):101290. doi: 10.1016/j.jceh.2023.10.001. Epub 2023 Oct 5. J Clin Exp Hepatol. 2024. PMID: 38544763 Free PMC article.
-
A Rare Case of Progressive Familial Intrahepatic Cholestasis Type 4: A Case Report and Literature Review.Cureus. 2023 Oct 18;15(10):e47276. doi: 10.7759/cureus.47276. eCollection 2023 Oct. Cureus. 2023. PMID: 38021987 Free PMC article.
-
NR1H4 mutation and rapid progressive intrahepatic cholestasis in infancy: A case report and literature review.Clin Case Rep. 2024 Feb 23;12(2):e8531. doi: 10.1002/ccr3.8531. eCollection 2024 Feb. Clin Case Rep. 2024. PMID: 38405357 Free PMC article.
-
Case Report: Add-on treatment with odevixibat in a new subtype of progressive familial intrahepatic cholestasis broadens the therapeutic horizon of genetic cholestasis.Front Pediatr. 2023 Feb 14;11:1061535. doi: 10.3389/fped.2023.1061535. eCollection 2023. Front Pediatr. 2023. PMID: 36865697 Free PMC article.
-
Consecutive baicalin treatment relieves its accumulation in rats with intrahepatic cholestasis by increasing MRP2 expression.Heliyon. 2023 Jan 3;9(1):e12689. doi: 10.1016/j.heliyon.2022.e12689. eCollection 2023 Jan. Heliyon. 2023. PMID: 36647350 Free PMC article.
References
-
- Hori T, Nguyen JH, Uemoto S. Progressive familial intrahepatic cholestasis. Hepatobiliary Pancreat Dis Int. 2010;9:570–578. - PubMed
-
- Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2012;36 Suppl 1:S26–S35. - PubMed
-
- Baker A, Kerkar N, Todorova L, Kamath BM, Houwen RHJ. Systematic review of progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2019;43:20–36. - PubMed
-
- Sambrotta M, Strautnieks S, Papouli E, Rushton P, Clark BE, Parry DA, Logan CV, Newbury LJ, Kamath BM, Ling S, Grammatikopoulos T, Wagner BE, Magee JC, Sokol RJ, Mieli-Vergani G University of Washington Center for Mendelian Genomics, Smith JD, Johnson CA, McClean P, Simpson MA, Knisely AS, Bull LN, Thompson RJ. Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet. 2014;46:326–328. - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
