

Evidence-Based Assessment of Genes in Dilated Cardiomyopathy
- PMID: 33947203
- PMCID: PMC8247549
- DOI: 10.1161/CIRCULATIONAHA.120.053033
Evidence-Based Assessment of Genes in Dilated Cardiomyopathy
Abstract
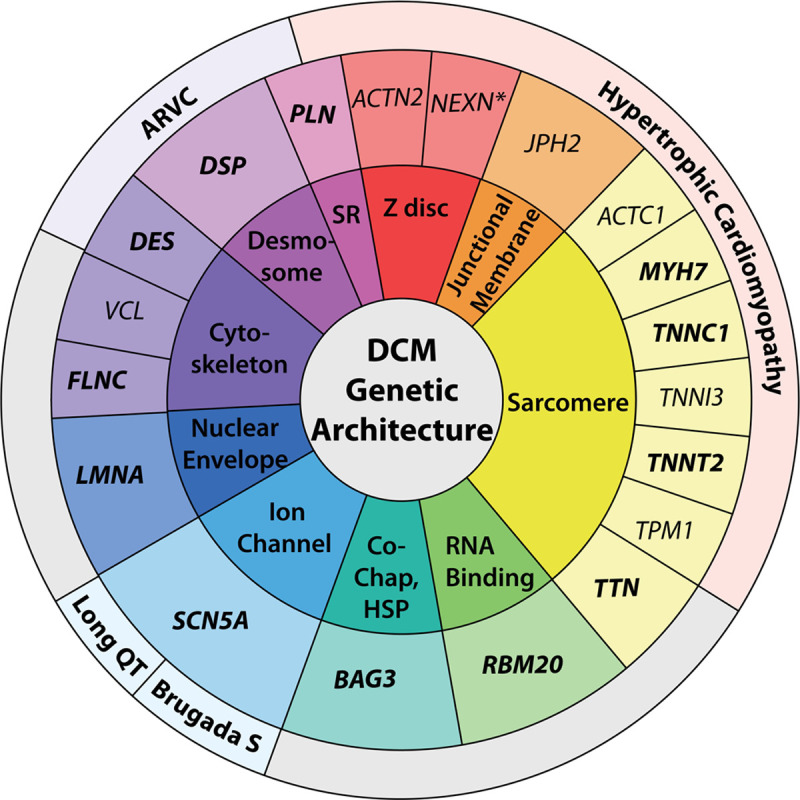
Background: Each of the cardiomyopathies, classically categorized as hypertrophic cardiomyopathy, dilated cardiomyopathy (DCM), and arrhythmogenic right ventricular cardiomyopathy, has a signature genetic theme. Hypertrophic cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy are largely understood as genetic diseases of sarcomere or desmosome proteins, respectively. In contrast, >250 genes spanning >10 gene ontologies have been implicated in DCM, representing a complex and diverse genetic architecture. To clarify this, a systematic curation of evidence to establish the relationship of genes with DCM was conducted.
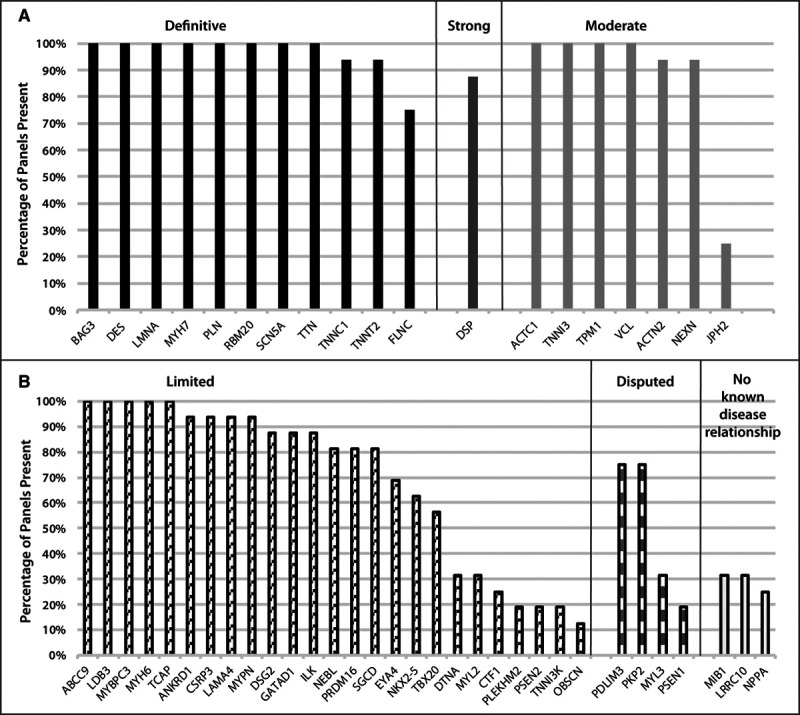
Methods: An international panel with clinical and scientific expertise in DCM genetics evaluated evidence supporting monogenic relationships of genes with idiopathic DCM. The panel used the Clinical Genome Resource semiquantitative gene-disease clinical validity classification framework with modifications for DCM genetics to classify genes into categories on the basis of the strength of currently available evidence. Representation of DCM genes on clinically available genetic testing panels was evaluated.
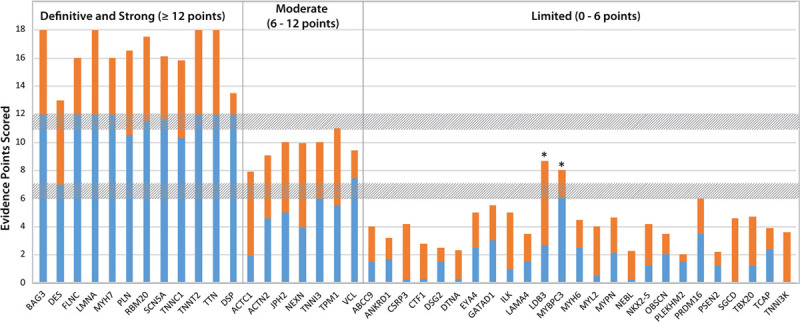
Results: Fifty-one genes with human genetic evidence were curated. Twelve genes (23%) from 8 gene ontologies were classified as having definitive (BAG3, DES, FLNC, LMNA, MYH7, PLN, RBM20, SCN5A, TNNC1, TNNT2, TTN) or strong (DSP) evidence. Seven genes (14%; ACTC1, ACTN2, JPH2, NEXN, TNNI3, TPM1, VCL) including 2 additional ontologies were classified as moderate evidence; these genes are likely to emerge as strong or definitive with additional evidence. Of these 19 genes, 6 were similarly classified for hypertrophic cardiomyopathy and 3 for arrhythmogenic right ventricular cardiomyopathy. Of the remaining 32 genes (63%), 25 (49%) had limited evidence, 4 (8%) were disputed, 2 (4%) had no disease relationship, and 1 (2%) was supported by animal model data only. Of the 16 evaluated clinical genetic testing panels, most definitive genes were included, but panels also included numerous genes with minimal human evidence.
Conclusions: In the curation of 51 genes, 19 had high evidence (12 definitive/strong, 7 moderate). It is notable that these 19 genes explain only a minority of cases, leaving the remainder of DCM genetic architecture incompletely addressed. Clinical genetic testing panels include most high-evidence genes; however, genes lacking robust evidence are also commonly included. We recommend that high-evidence DCM genes be used for clinical practice and that caution be exercised in the interpretation of variants in variable-evidence DCM genes.
Keywords: cardiomyopathy; genetics.
Figures
Comment in
-
Reappraising Genes for Dilated Cardiomyopathy: Stepping Back to Move Forward.Circulation. 2021 Jul 6;144(1):20-22. doi: 10.1161/CIRCULATIONAHA.121.054961. Epub 2021 Jul 6. Circulation. 2021. PMID: 34228479 No abstract available.
Similar articles
-
Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy.Circulation. 2020 Feb 4;141(5):387-398. doi: 10.1161/CIRCULATIONAHA.119.037661. Epub 2020 Jan 27. Circulation. 2020. PMID: 31983221 Free PMC article.
-
Targeted next-generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy.Int J Mol Med. 2015 Dec;36(6):1479-86. doi: 10.3892/ijmm.2015.2361. Epub 2015 Oct 7. Int J Mol Med. 2015. PMID: 26458567 Free PMC article.
-
Dilated Cardiomyopathy Overview.2007 Jul 27 [updated 2024 Dec 12]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2007 Jul 27 [updated 2024 Dec 12]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301486 Free Books & Documents. Review.
-
Emerging concepts in arrhythmogenic dilated cardiomyopathy.Heart Fail Rev. 2021 Sep;26(5):1219-1229. doi: 10.1007/s10741-020-09933-z. Heart Fail Rev. 2021. PMID: 32056050 Review.
-
Genetic testing for dilated cardiomyopathy in clinical practice.J Card Fail. 2012 Apr;18(4):296-303. doi: 10.1016/j.cardfail.2012.01.013. Epub 2012 Feb 15. J Card Fail. 2012. PMID: 22464770 Free PMC article.
Cited by
-
The Complex and Diverse Genetic Architecture of Dilated Cardiomyopathy.Circ Res. 2021 May 14;128(10):1514-1532. doi: 10.1161/CIRCRESAHA.121.318157. Epub 2021 May 13. Circ Res. 2021. PMID: 33983834 Free PMC article. Review.
-
Allelic heterogeneity of TTNtv dilated cardiomyopathy can be modeled in adult zebrafish.JCI Insight. 2024 Apr 8;9(7):e175501. doi: 10.1172/jci.insight.175501. JCI Insight. 2024. PMID: 38412038 Free PMC article.
-
Genetic contributions to risk of adverse pregnancy outcomes.Curr Cardiovasc Risk Rep. 2023 Nov;17(11):185-193. doi: 10.1007/s12170-023-00729-y. Epub 2023 Sep 28. Curr Cardiovasc Risk Rep. 2023. PMID: 38186860 Free PMC article.
-
The genetic basis for adult-onset idiopathic dilated cardiomyopathy in people of African descent.Heart Fail Rev. 2023 Jul;28(4):879-892. doi: 10.1007/s10741-023-10302-9. Epub 2023 Mar 14. Heart Fail Rev. 2023. PMID: 36917398 Free PMC article. Review.
-
The role of genetic testing in diagnosis and care of inherited cardiac conditions in a specialised multidisciplinary clinic.Genome Med. 2022 Dec 28;14(1):145. doi: 10.1186/s13073-022-01149-0. Genome Med. 2022. PMID: 36578016 Free PMC article.
References
-
- Falk RH, Hershberger RE. Zipes DP, Libby P, Bonow RO, Mann DL, Tomaselli G, eds. The dilated, restrictive, and infiltrative cardiomyopathies. In: Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. 2018. 11th Edition. Elsevier
-
- Jarcho JA, McKenna W, Pare JA, Solomon SD, Holcombe RF, Dickie S, Levi T, Donis-Keller H, Seidman JG, Seidman CE. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med. 1989;321:1372–1378. doi: 10.1056/NEJM198911163212005 - PubMed
-
- Graber HL, Unverferth DV, Baker PB, Ryan JM, Baba N, Wooley CF. Evolution of a hereditary cardiac conduction and muscle disorder: a study involving a family with six generations affected. Circulation. 1986;74:21–35. doi: 10.1161/01.cir.74.1.21 - PubMed
-
- Nava A, Bauce B, Basso C, Muriago M, Rampazzo A, Villanova C, Daliento L, Buja G, Corrado D, Danieli GA, et al. . Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36:2226–2233. doi: 10.1016/s0735-1097(00)00997-9 - PubMed
-
- Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
