

Defining the clinical phenotype of Saul-Wilson syndrome
- PMID: 31949312
- PMCID: PMC7205587
- DOI: 10.1038/s41436-019-0737-1
Defining the clinical phenotype of Saul-Wilson syndrome
Abstract
Purpose: Four patients with Saul-Wilson syndrome were reported between 1982 and 1994, but no additional individuals were described until 2018, when the molecular etiology of the disease was elucidated. Hence, the clinical phenotype of the disease remains poorly defined. We address this shortcoming by providing a detailed characterization of its phenotype.
Methods: Retrospective chart reviews were performed and primary radiographs assessed for all 14 individuals. Four individuals underwent detailed ophthalmologic examination by the same physician. Two individuals underwent gynecologic evaluation. Z-scores for height, weight, head circumference and body mass index were calculated at different ages.
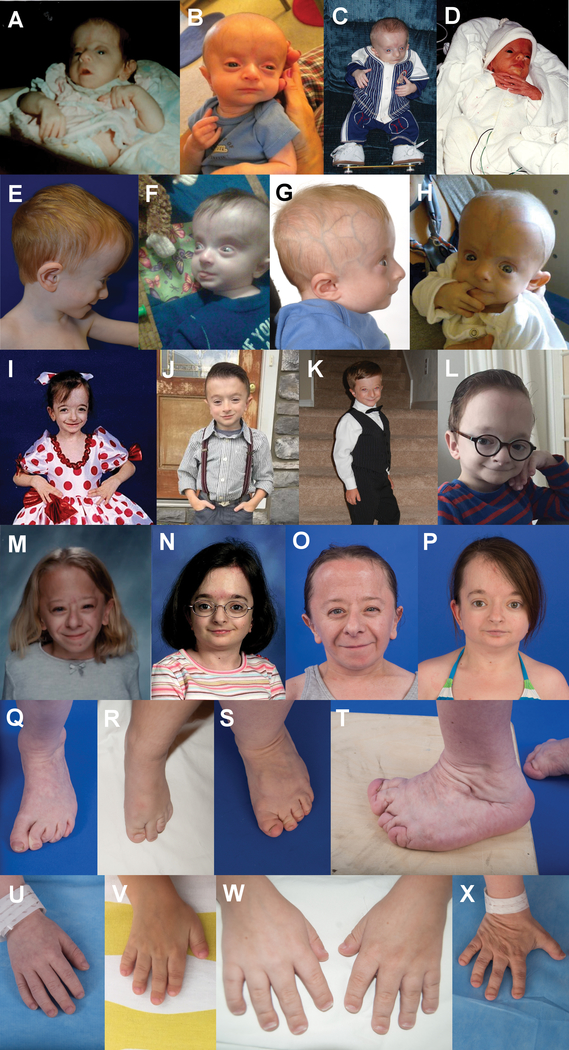
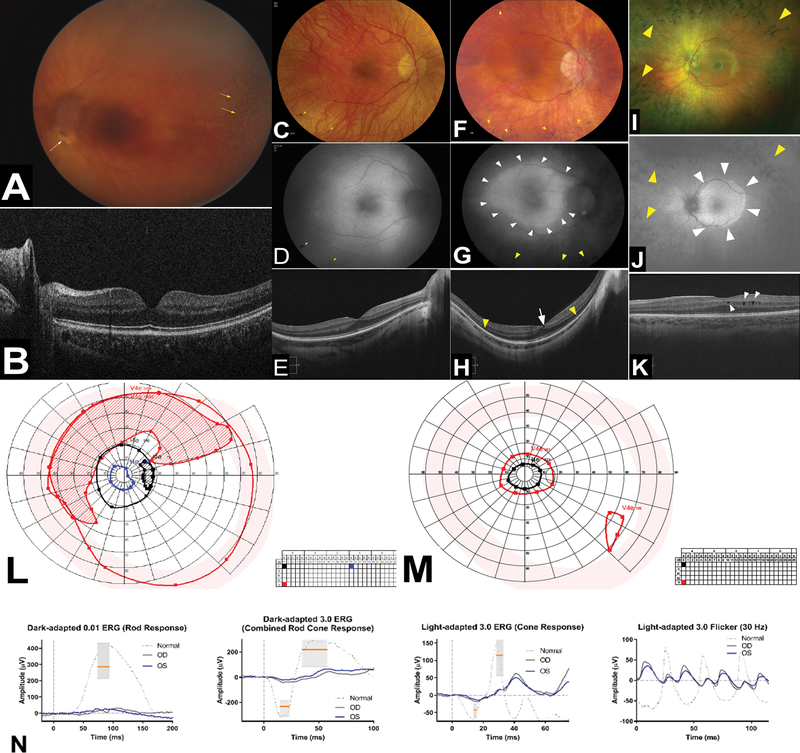
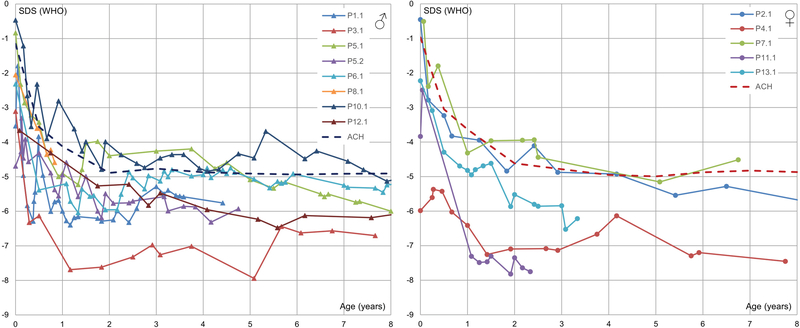
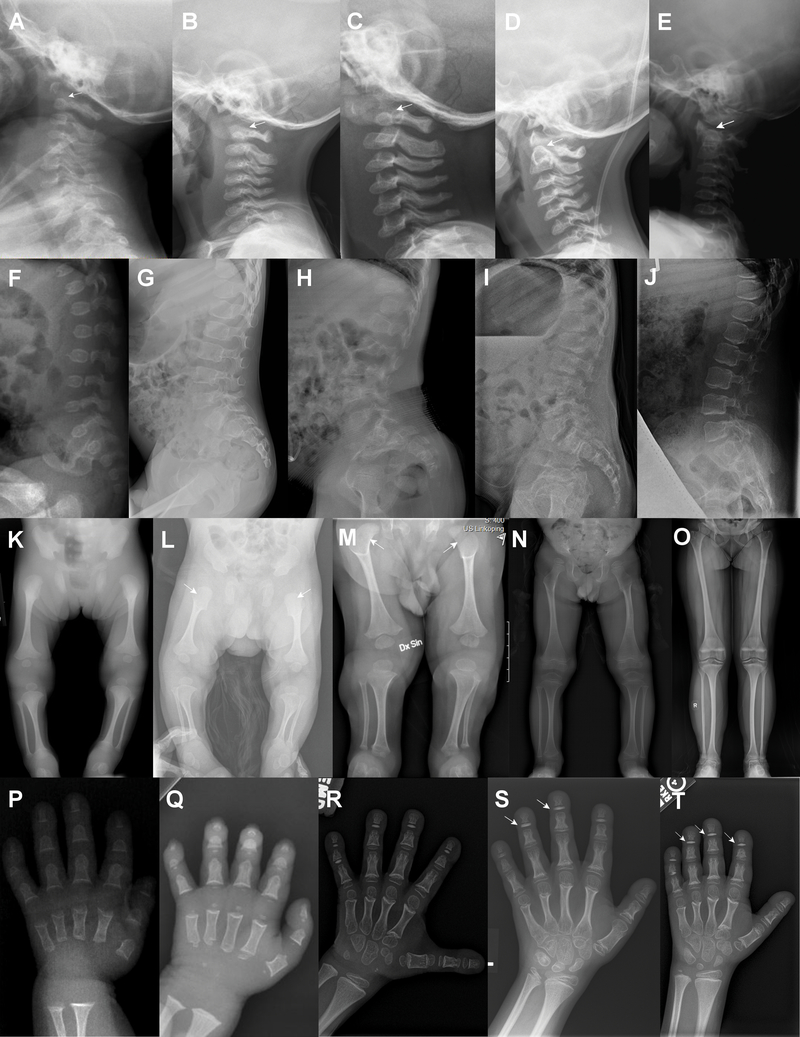
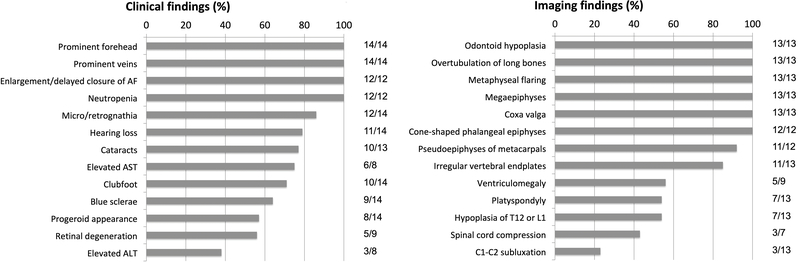
Results: All patients exhibited short stature, with sharp decline from the mean within the first months of life, and a final height Z-score between -4 and -8.5 standard deviations. The facial and radiographic features evolved over time. Intermittent neutropenia was frequently observed. Novel findings included elevation of liver transaminases, skeletal fragility, rod-cone dystrophy, and cystic macular changes.
Conclusions: Saul-Wilson syndrome presents a remarkably uniform phenotype, and the comprehensive description of our cohort allows for improved understanding of the long-term morbidity of the condition, establishment of follow-up recommendations for affected individuals, and documentation of the natural history into adulthood for comparison with treated patients, when therapeutics become available.
Keywords: COG4; G516R; Saul–Wilson syndrome; phenotype.
Conflict of interest statement
Conflict of Interest:
The authors declare no conflict of interest.
Figures
Similar articles
-
COG4 mutation in Saul-Wilson syndrome selectively affects secretion of proteins involved in chondrogenesis in chondrocyte-like cells.Front Cell Dev Biol. 2022 Oct 28;10:979096. doi: 10.3389/fcell.2022.979096. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36393834 Free PMC article.
-
Growth in individuals with Saul-Wilson syndrome.Am J Med Genet A. 2020 Sep;182(9):2110-2116. doi: 10.1002/ajmg.a.61754. Epub 2020 Jul 11. Am J Med Genet A. 2020. PMID: 32652690 Free PMC article.
-
A Dominant Heterozygous Mutation in COG4 Causes Saul-Wilson Syndrome, a Primordial Dwarfism, and Disrupts Zebrafish Development via Wnt Signaling.Front Cell Dev Biol. 2021 Sep 14;9:720688. doi: 10.3389/fcell.2021.720688. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34595172 Free PMC article.
-
Genetic short stature.Growth Horm IGF Res. 2018 Feb;38:29-33. doi: 10.1016/j.ghir.2017.12.003. Epub 2017 Dec 6. Growth Horm IGF Res. 2018. PMID: 29249624 Review.
-
Recombinant growth hormone in children and adolescents with Turner syndrome.Cochrane Database Syst Rev. 2003;(3):CD003887. doi: 10.1002/14651858.CD003887. Cochrane Database Syst Rev. 2003. Update in: Cochrane Database Syst Rev. 2007 Jan 24;(1):CD003887. doi: 10.1002/14651858.CD003887.pub2. PMID: 12917993 Updated. Review.
Cited by
-
COPB2 loss of function causes a coatopathy with osteoporosis and developmental delay.Am J Hum Genet. 2021 Sep 2;108(9):1710-1724. doi: 10.1016/j.ajhg.2021.08.002. Epub 2021 Aug 26. Am J Hum Genet. 2021. PMID: 34450031 Free PMC article.
-
Sequenced-based GWAS for linear classification traits in Belgian Blue beef cattle reveals new coding variants in genes regulating body size in mammals.Genet Sel Evol. 2023 Nov 28;55(1):83. doi: 10.1186/s12711-023-00857-4. Genet Sel Evol. 2023. PMID: 38017417 Free PMC article.
-
COG4 mutation in Saul-Wilson syndrome selectively affects secretion of proteins involved in chondrogenesis in chondrocyte-like cells.Front Cell Dev Biol. 2022 Oct 28;10:979096. doi: 10.3389/fcell.2022.979096. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36393834 Free PMC article.
-
Clinical and biochemical footprints of congenital disorders of glycosylation: Proposed nosology.Mol Genet Metab. 2024 May;142(1):108476. doi: 10.1016/j.ymgme.2024.108476. Epub 2024 Apr 10. Mol Genet Metab. 2024. PMID: 38653092 Free PMC article. Review.
-
Craniofacial disorders and dysplasias: Molecular, clinical, and management perspectives.Bone Rep. 2024 Mar 1;20:101747. doi: 10.1016/j.bonr.2024.101747. eCollection 2024 Mar. Bone Rep. 2024. PMID: 38566929 Free PMC article. Review.
References
-
- Saul RA. Unknown cases. Proceedings of the Greenwood Genetic Center. 1982;1(1):102–105.
-
- Saul RA, Wilson WG. A “new” skeletal dysplasia in two unrelated boys. Am J Med Genet. 1990;35(3):388–393. - PubMed
-
- Hersh JH, Joyce MR, Spranger J, et al. Microcephalic osteodysplastic dysplasia. Am J Med Genet. 1994;51(3):194–199. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
