

Autozygome and high throughput confirmation of disease genes candidacy
- PMID: 30237576
- PMCID: PMC6752307
- DOI: 10.1038/s41436-018-0138-x
Autozygome and high throughput confirmation of disease genes candidacy
Abstract
Purpose: Establishing links between Mendelian phenotypes and genes enables the proper interpretation of variants therein. Autozygome, a rich source of homozygous variants, has been successfully utilized for the high throughput identification of novel autosomal recessive disease genes. Here, we highlight the utility of the autozygome for the high throughput confirmation of previously published tentative links to diseases.
Methods: Autozygome and exome analysis of patients with suspected Mendelian phenotypes. All variants were classified according to the American College of Medical Genetics and Genomics guidelines.
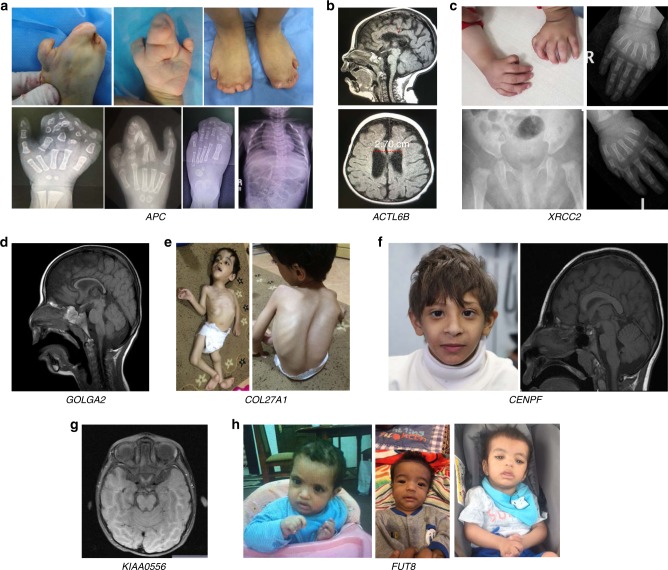
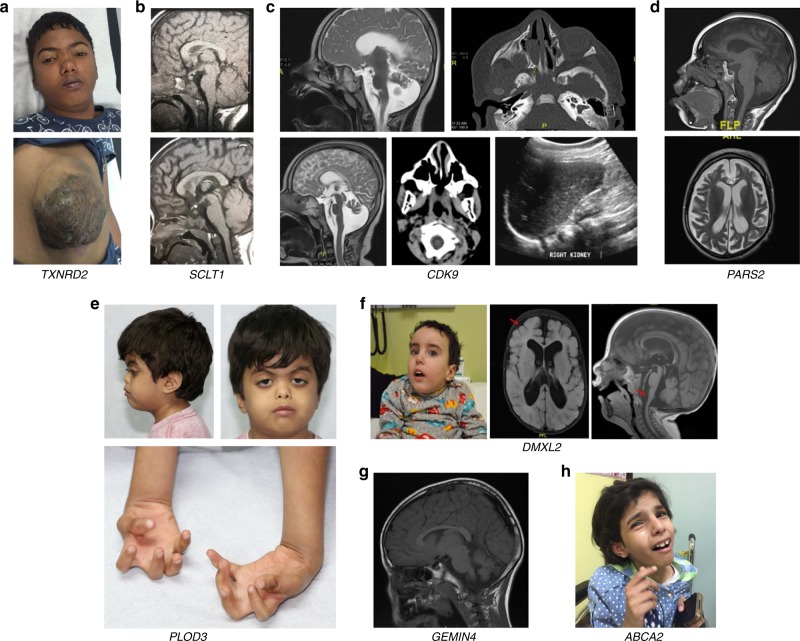
Results: We highlight 30 published candidate genes (ACTL6B, ADAM22, AGTPBP1, APC, C12orf4, C3orf17 (NEPRO), CENPF, CNPY3, COL27A1, DMBX1, FUT8, GOLGA2, KIAA0556, LENG8, MCIDAS, MTMR9, MYH11, QRSL1, RUBCN, SLC25A42, SLC9A1, TBXT, TFG, THUMPD1, TRAF3IP2, UFC1, UFM1, WDR81, XRCC2, ZAK) in which we identified homozygous likely deleterious variants in patients with compatible phenotypes. We also identified homozygous likely deleterious variants in 18 published candidate genes (ABCA2, ARL6IP1, ATP8A2, CDK9, CNKSR1, DGAT1, DMXL2, GEMIN4, HCN2, HCRT, MYO9A, PARS2, PLOD3, PREPL, SCLT1, STX3, TXNRD2, WIPI2) although the associated phenotypes are sufficiently different from the original reports that they represent phenotypic expansion or potentially distinct allelic disorders.
Conclusions: Our results should facilitate the timely relabeling of these candidate disease genes in relevant databases to improve the yield of clinical genomic sequencing.
Keywords: ACMG guidelines; candidate genes; variant interpretation.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population.Am J Hum Genet. 2019 Jun 6;104(6):1182-1201. doi: 10.1016/j.ajhg.2019.04.011. Epub 2019 May 23. Am J Hum Genet. 2019. PMID: 31130284 Free PMC article.
-
Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium.Am J Hum Genet. 2016 Jun 2;98(6):1067-1076. doi: 10.1016/j.ajhg.2016.03.024. Epub 2016 May 12. Am J Hum Genet. 2016. PMID: 27181684 Free PMC article.
-
Accelerating matchmaking of novel dysmorphology syndromes through clinical and genomic characterization of a large cohort.Genet Med. 2016 Jul;18(7):686-95. doi: 10.1038/gim.2015.147. Epub 2015 Dec 3. Genet Med. 2016. PMID: 26633546
-
A retrospective review of multiple findings in diagnostic exome sequencing: half are distinct and half are overlapping diagnoses.Genet Med. 2019 Oct;21(10):2199-2207. doi: 10.1038/s41436-019-0477-2. Epub 2019 Mar 21. Genet Med. 2019. PMID: 30894705 Free PMC article. Review.
-
Navigating the nuances of clinical sequence variant interpretation in Mendelian disease.Genet Med. 2018 Sep;20(9):918-926. doi: 10.1038/s41436-018-0100-y. Epub 2018 Jul 10. Genet Med. 2018. PMID: 29988079 Free PMC article. Review.
Cited by
-
Twenty Novel Disease Group-Specific and 12 New Shared Macrophage Pathways in Eight Groups of 34 Diseases Including 24 Inflammatory Organ Diseases and 10 Types of Tumors.Front Immunol. 2019 Nov 14;10:2612. doi: 10.3389/fimmu.2019.02612. eCollection 2019. Front Immunol. 2019. PMID: 31824480 Free PMC article.
-
A Syrian patient with Steel syndrome due to compound heterozygous COL27A1 mutations with colobomata of the eye.Am J Med Genet A. 2020 Apr;182(4):730-734. doi: 10.1002/ajmg.a.61478. Epub 2020 Jan 8. Am J Med Genet A. 2020. PMID: 31913554 Free PMC article.
-
Mutations in PIGB Cause an Inherited GPI Biosynthesis Defect with an Axonal Neuropathy and Metabolic Abnormality in Severe Cases.Am J Hum Genet. 2019 Aug 1;105(2):384-394. doi: 10.1016/j.ajhg.2019.05.019. Epub 2019 Jun 27. Am J Hum Genet. 2019. PMID: 31256876 Free PMC article.
-
PARS2-associated mitochondrial disease: A case report of a patient with prolonged survival and literature review.Mol Genet Metab Rep. 2020 Jun 2;24:100613. doi: 10.1016/j.ymgmr.2020.100613. eCollection 2020 Sep. Mol Genet Metab Rep. 2020. PMID: 32514400 Free PMC article.
-
Autophagy genes in biology and disease.Nat Rev Genet. 2023 Jun;24(6):382-400. doi: 10.1038/s41576-022-00562-w. Epub 2023 Jan 12. Nat Rev Genet. 2023. PMID: 36635405 Free PMC article. Review.
References
-
- Wright Caroline F, McRae Jeremy F, Clayton Stephen, Gallone Giuseppe, Aitken Stuart, FitzGerald Tomas W, Jones Philip, Prigmore Elena, Rajan Diana, Lord Jenny, Sifrim Alejandro, Kelsell Rosemary, Parker Michael J, Barrett Jeffrey C, Hurles Matthew E, FitzPatrick David R, Firth Helen V. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genetics in Medicine. 2018;20(10):1216–1223. doi: 10.1038/gim.2017.246. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
