

The Neuromuscular Junction and Wide Heterogeneity of Congenital Myasthenic Syndromes
- PMID: 29874875
- PMCID: PMC6032286
- DOI: 10.3390/ijms19061677
The Neuromuscular Junction and Wide Heterogeneity of Congenital Myasthenic Syndromes
Abstract
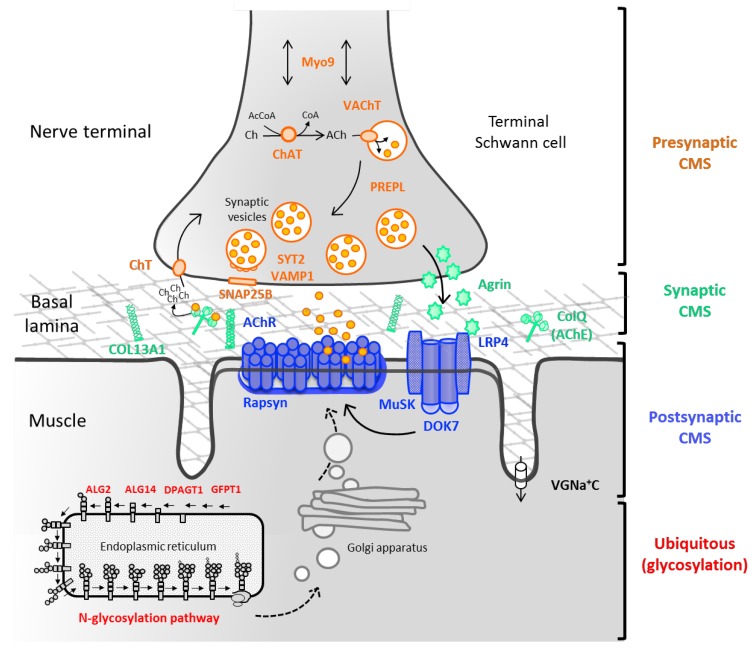
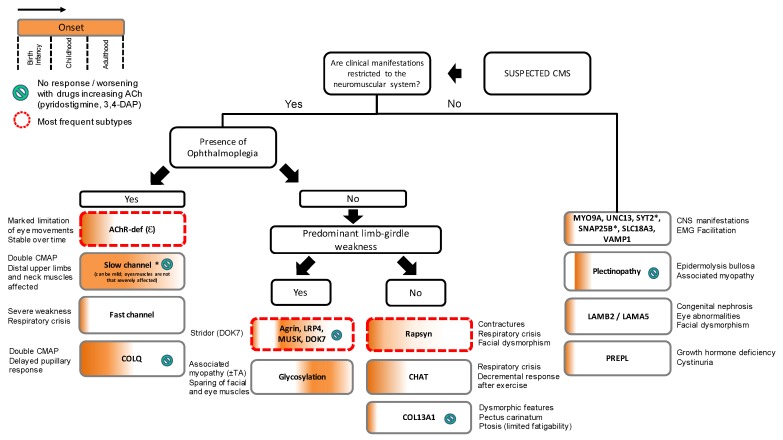
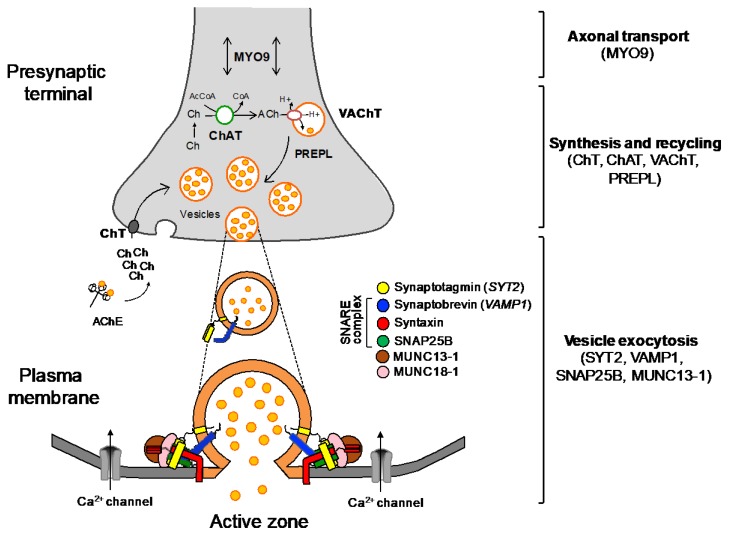
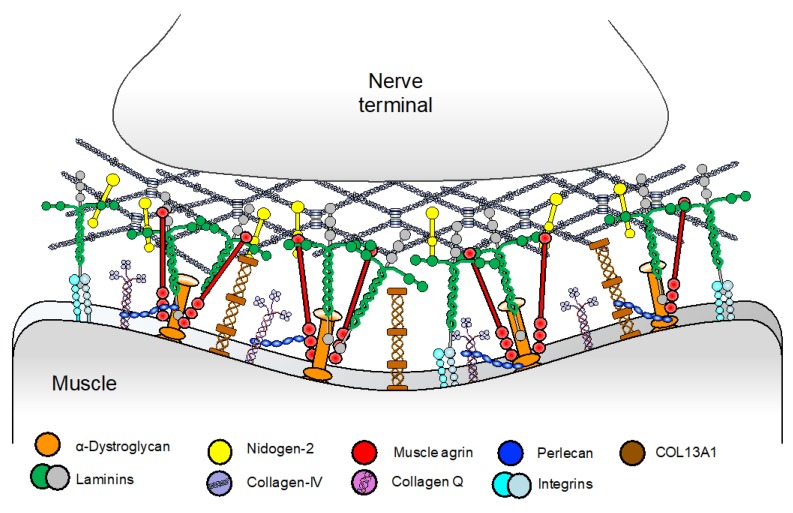
Congenital myasthenic syndromes (CMS) are genetic disorders characterised by impaired neuromuscular transmission. This review provides an overview on CMS and highlights recent advances in the field, including novel CMS causative genes and improved therapeutic strategies. CMS due to mutations in SLC5A7 and SLC18A3, impairing the synthesis and recycling of acetylcholine, have recently been described. In addition, a novel group of CMS due to mutations in SNAP25B, SYT2, VAMP1, and UNC13A1 encoding molecules implicated in synaptic vesicles exocytosis has been characterised. The increasing number of presynaptic CMS exhibiting CNS manifestations along with neuromuscular weakness demonstrate that the myasthenia can be only a small part of a much more extensive disease phenotype. Moreover, the spectrum of glycosylation abnormalities has been increased with the report that GMPPB mutations can cause CMS, thus bridging myasthenic disorders with dystroglycanopathies. Finally, the discovery of COL13A1 mutations and laminin α5 deficiency has helped to draw attention to the role of extracellular matrix proteins for the formation and maintenance of muscle endplates. The benefit of β2-adrenergic agonists alone or combined with pyridostigmine or 3,4-Dyaminopiridine is increasingly being reported for different subtypes of CMS including AChR-deficiency and glycosylation abnormalities, thus expanding the therapeutic repertoire available.
Keywords: COL13A1; GMPPB; N-glycosylation pathway; SNARE complex; congenital myasthenic syndromes; neuromuscular junction; neuromuscular transmission; presynaptic CMS; β2-adrenergic agonists.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Congenital myasthenic syndromes: recent advances.Curr Opin Neurol. 2016 Oct;29(5):565-71. doi: 10.1097/WCO.0000000000000370. Curr Opin Neurol. 2016. PMID: 27472506 Review.
-
[Congenital myasthenic syndromes: phenotypic expression and pathophysiological characterisation].Rev Neurol (Paris). 2004 Feb;160(2):163-76. doi: 10.1016/s0035-3787(04)70887-5. Rev Neurol (Paris). 2004. PMID: 15034473 Review. French.
-
Genetic heterogeneity and pathophysiological mechanisms in congenital myasthenic syndromes.Eur J Paediatr Neurol. 2011 May;15(3):189-96. doi: 10.1016/j.ejpn.2011.03.006. Epub 2011 Apr 17. Eur J Paediatr Neurol. 2011. PMID: 21498094 Review.
-
Congenital myasthenic syndromes and the neuromuscular junction.Curr Opin Neurol. 2014 Oct;27(5):566-75. doi: 10.1097/WCO.0000000000000134. Curr Opin Neurol. 2014. PMID: 25159927 Review.
-
Clinical and Pathologic Features of Congenital Myasthenic Syndromes Caused by 35 Genes-A Comprehensive Review.Int J Mol Sci. 2023 Feb 13;24(4):3730. doi: 10.3390/ijms24043730. Int J Mol Sci. 2023. PMID: 36835142 Free PMC article. Review.
Cited by
-
A Pediatric Case of COLQ-Related Congenital Myasthenic Syndrome with Marked Fatigue.Children (Basel). 2023 Apr 24;10(5):769. doi: 10.3390/children10050769. Children (Basel). 2023. PMID: 37238317 Free PMC article.
-
Congenital myasthenic syndrome in China: genetic and myopathological characterization.Ann Clin Transl Neurol. 2021 Apr;8(4):898-907. doi: 10.1002/acn3.51346. Epub 2021 Mar 23. Ann Clin Transl Neurol. 2021. PMID: 33756069 Free PMC article.
-
Muscle-Specific Kinase Myasthenia Gravis.Front Immunol. 2020 May 8;11:707. doi: 10.3389/fimmu.2020.00707. eCollection 2020. Front Immunol. 2020. PMID: 32457737 Free PMC article. Review.
-
New mutation in the β1 propeller domain of LRP4 responsible for congenital myasthenic syndrome associated with Cenani-Lenz syndrome.Sci Rep. 2023 Aug 28;13(1):14054. doi: 10.1038/s41598-023-41008-5. Sci Rep. 2023. PMID: 37640745 Free PMC article.
-
Myasthenia Gravis: Pathogenic Effects of Autoantibodies on Neuromuscular Architecture.Cells. 2019 Jul 2;8(7):671. doi: 10.3390/cells8070671. Cells. 2019. PMID: 31269763 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
