

Outlier detection in BLAST hits
- PMID: 29588650
- PMCID: PMC5863388
- DOI: 10.1186/s13015-018-0126-3
Outlier detection in BLAST hits
Abstract
Background: An important task in a metagenomic analysis is the assignment of taxonomic labels to sequences in a sample. Most widely used methods for taxonomy assignment compare a sequence in the sample to a database of known sequences. Many approaches use the best BLAST hit(s) to assign the taxonomic label. However, it is known that the best BLAST hit may not always correspond to the best taxonomic match. An alternative approach involves phylogenetic methods, which take into account alignments and a model of evolution in order to more accurately define the taxonomic origin of sequences. Similarity-search based methods typically run faster than phylogenetic methods and work well when the organisms in the sample are well represented in the database. In contrast, phylogenetic methods have the capability to identify new organisms in a sample but are computationally quite expensive.
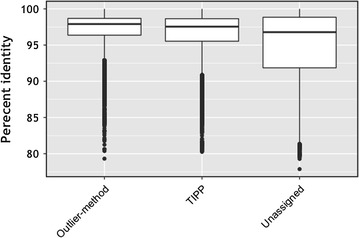
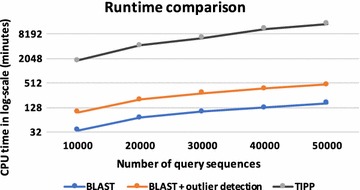
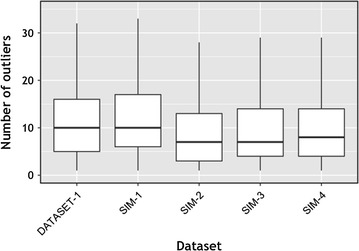
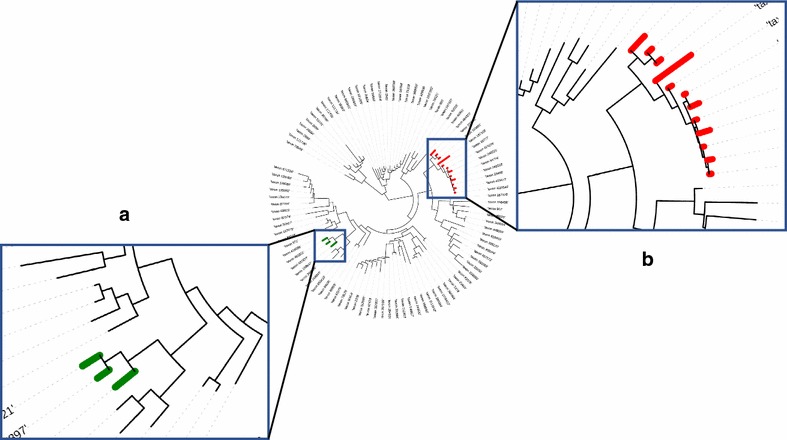
Results: We propose a two-step approach for metagenomic taxon identification; i.e., use a rapid method that accurately classifies sequences using a reference database (this is a filtering step) and then use a more complex phylogenetic method for the sequences that were unclassified in the previous step. In this work, we explore whether and when using top BLAST hit(s) yields a correct taxonomic label. We develop a method to detect outliers among BLAST hits in order to separate the phylogenetically most closely related matches from matches to sequences from more distantly related organisms. We used modified BILD (Bayesian Integral Log-Odds) scores, a multiple-alignment scoring function, to define the outliers within a subset of top BLAST hits and assign taxonomic labels. We compared the accuracy of our method to the RDP classifier and show that our method yields fewer misclassifications while properly classifying organisms that are not present in the database. Finally, we evaluated the use of our method as a pre-processing step before more expensive phylogenetic analyses (in our case TIPP) in the context of real 16S rRNA datasets.
Conclusion: Our experiments make a good case for using a two-step approach for accurate taxonomic assignment. We show that our method can be used as a filtering step before using phylogenetic methods and provides a way to interpret BLAST results using more information than provided by E-values and bit-scores alone.
Keywords: Metagenomics; Outlier detection; Sequence alignment; Taxonomy classification.
Figures
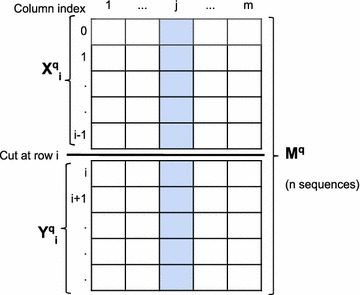
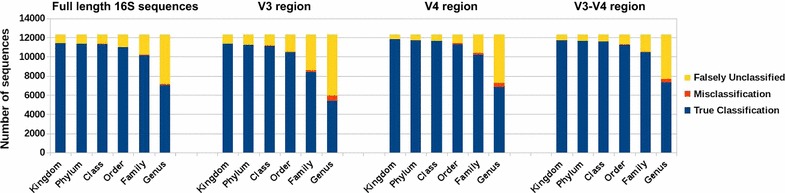
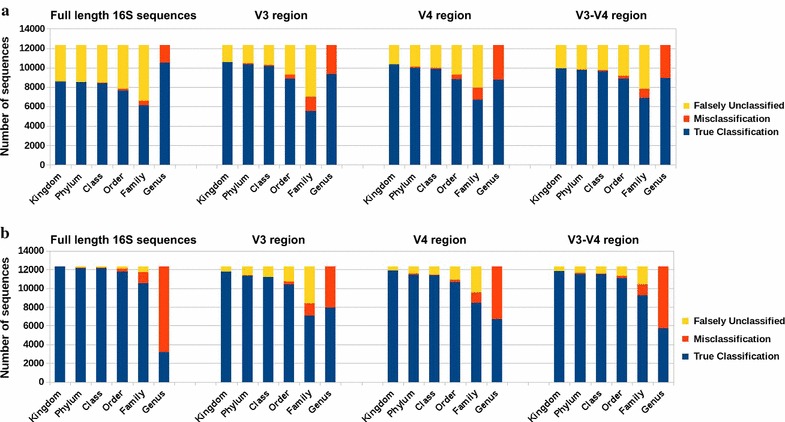
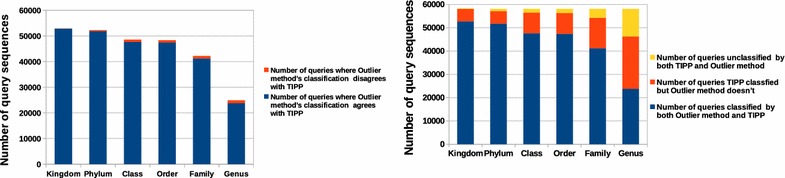

Similar articles
-
How reliable is metabarcoding for pollen identification? An evaluation of different taxonomic assignment strategies by cross-validation.PeerJ. 2024 Jan 31;12:e16567. doi: 10.7717/peerj.16567. eCollection 2024. PeerJ. 2024. PMID: 38313030 Free PMC article.
-
A Bayesian taxonomic classification method for 16S rRNA gene sequences with improved species-level accuracy.BMC Bioinformatics. 2017 May 10;18(1):247. doi: 10.1186/s12859-017-1670-4. BMC Bioinformatics. 2017. PMID: 28486927 Free PMC article.
-
TaxAss: Leveraging a Custom Freshwater Database Achieves Fine-Scale Taxonomic Resolution.mSphere. 2018 Sep 5;3(5):e00327-18. doi: 10.1128/mSphere.00327-18. mSphere. 2018. PMID: 30185512 Free PMC article.
-
Metagenomic Analysis Using Phylogenetic Placement-A Review of the First Decade.Front Bioinform. 2022 May 26;2:871393. doi: 10.3389/fbinf.2022.871393. eCollection 2022. Front Bioinform. 2022. PMID: 36304302 Free PMC article. Review.
-
Reference databases for taxonomic assignment in metagenomics.Brief Bioinform. 2012 Nov;13(6):682-95. doi: 10.1093/bib/bbs036. Epub 2012 Jul 10. Brief Bioinform. 2012. PMID: 22786784 Review.
Cited by
-
Metagenome reveals the midgut microbial community of Haemaphysalis qinghaiensis ticks collected from yaks and Tibetan sheep.Parasit Vectors. 2024 Aug 31;17(1):370. doi: 10.1186/s13071-024-06442-y. Parasit Vectors. 2024. PMID: 39217389 Free PMC article.
-
Embracing Ambiguity in the Taxonomic Classification of Microbiome Sequencing Data.Front Genet. 2019 Oct 17;10:1022. doi: 10.3389/fgene.2019.01022. eCollection 2019. Front Genet. 2019. PMID: 31681437 Free PMC article.
-
Viruses of Polar Aquatic Environments.Viruses. 2019 Feb 22;11(2):189. doi: 10.3390/v11020189. Viruses. 2019. PMID: 30813316 Free PMC article. Review.
-
SeqScreen: accurate and sensitive functional screening of pathogenic sequences via ensemble learning.Genome Biol. 2022 Jun 20;23(1):133. doi: 10.1186/s13059-022-02695-x. Genome Biol. 2022. PMID: 35725628 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
