

Natural history of infantile-onset spinal muscular atrophy
- PMID: 29149772
- PMCID: PMC5776712
- DOI: 10.1002/ana.25101
Natural history of infantile-onset spinal muscular atrophy
Abstract
Objective: Infantile-onset spinal muscular atrophy (SMA) is the most common genetic cause of infant mortality, typically resulting in death preceding age 2. Clinical trials in this population require an understanding of disease progression and identification of meaningful biomarkers to hasten therapeutic development and predict outcomes.
Methods: A longitudinal, multicenter, prospective natural history study enrolled 26 SMA infants and 27 control infants aged <6 months. Recruitment occurred at 14 centers over 21 months within the NINDS-sponsored NeuroNEXT (National Network for Excellence in Neuroscience Clinical Trials) Network. Infant motor function scales (Test of Infant Motor Performance Screening Items [TIMPSI], The Children's Hospital of Philadelphia Infant Test for Neuromuscular Disorders, and Alberta Infant Motor Score) and putative physiological and molecular biomarkers were assessed preceding age 6 months and at 6, 9, 12, 18, and 24 months with progression, correlations between motor function and biomarkers, and hazard ratios analyzed.
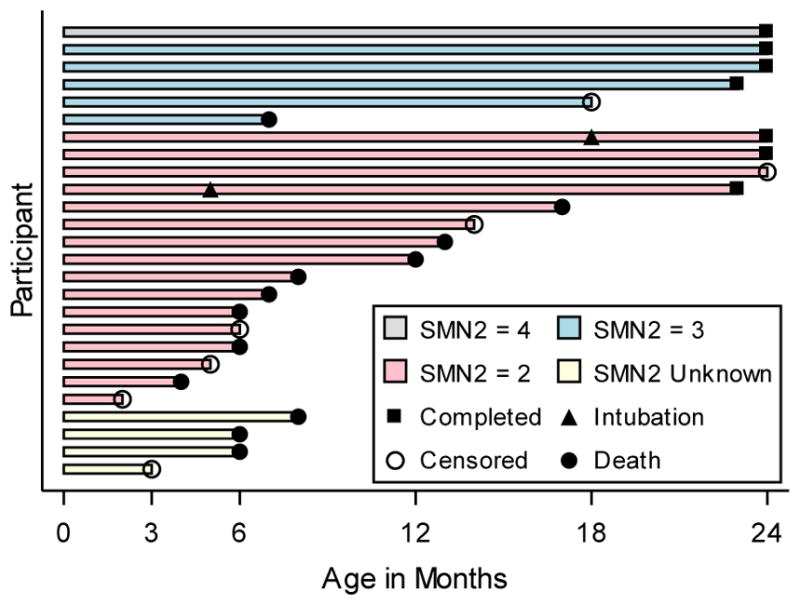
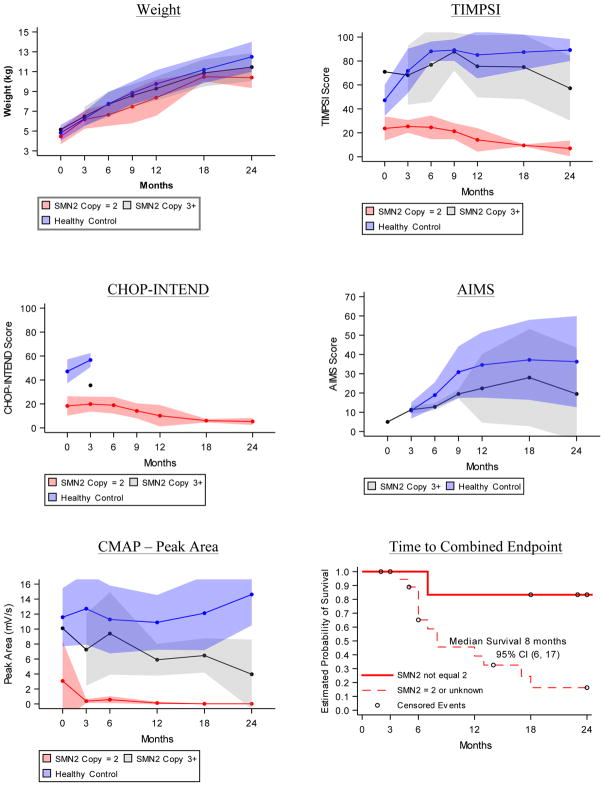
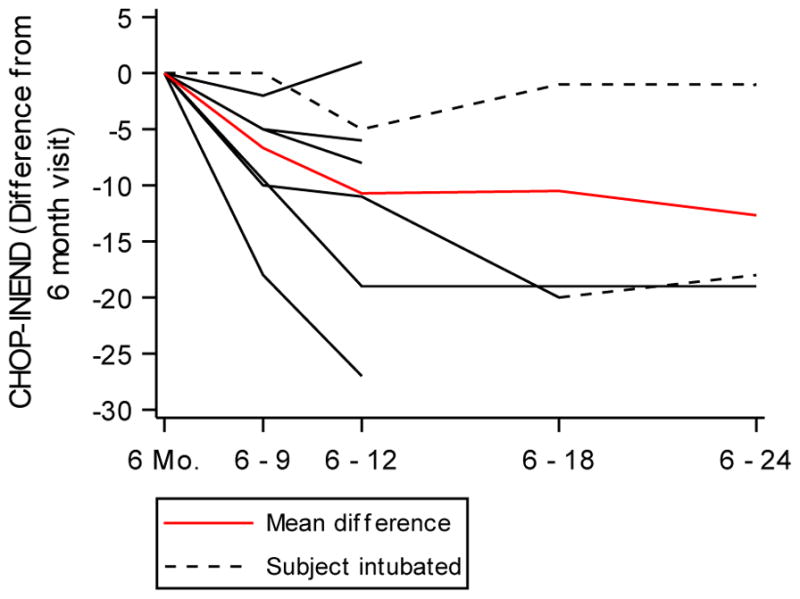
Results: Motor function scores (MFS) and compound muscle action potential (CMAP) decreased rapidly in SMA infants, whereas MFS in all healthy infants rapidly increased. Correlations were identified between TIMPSI and CMAP in SMA infants. TIMPSI at first study visit was associated with risk of combined endpoint of death or permanent invasive ventilation in SMA infants. Post-hoc analysis of survival to combined endpoint in SMA infants with 2 copies of SMN2 indicated a median age of 8 months at death (95% confidence interval, 6, 17).
Interpretation: These data of SMA and control outcome measures delineates meaningful change in clinical trials in infantile-onset SMA. The power and utility of NeuroNEXT to provide "real-world," prospective natural history data sets to accelerate public and private drug development programs for rare disease is demonstrated. Ann Neurol 2017;82:883-891.
© 2017 American Neurological Association.
Conflict of interest statement
The authors have nothing to report.
Figures
Comment in
-
NeuroNEXT is at your service.Ann Neurol. 2017 Dec;82(6):857-858. doi: 10.1002/ana.25100. Epub 2017 Dec 4. Ann Neurol. 2017. PMID: 29149769 No abstract available.
-
Building on NeuroNEXT: Next generation clinics to cure chronic neurological disability.Ann Neurol. 2017 Dec;82(6):859-862. doi: 10.1002/ana.25108. Epub 2017 Dec 8. Ann Neurol. 2017. PMID: 29171907 Free PMC article. No abstract available.
Similar articles
-
Baseline results of the NeuroNEXT spinal muscular atrophy infant biomarker study.Ann Clin Transl Neurol. 2016 Jan 21;3(2):132-45. doi: 10.1002/acn3.283. eCollection 2016 Feb. Ann Clin Transl Neurol. 2016. PMID: 26900585 Free PMC article.
-
Motor Function Test Reliability During the NeuroNEXT Spinal Muscular Atrophy Infant Biomarker Study.J Neuromuscul Dis. 2018;5(4):509-521. doi: 10.3233/JND-180327. J Neuromuscul Dis. 2018. PMID: 30223401 Free PMC article.
-
Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial.Lancet Neurol. 2021 Apr;20(4):284-293. doi: 10.1016/S1474-4422(21)00001-6. Epub 2021 Mar 17. Lancet Neurol. 2021. PMID: 33743238 Clinical Trial.
-
[Spinal muscular atrophy : Time for newborn screening?].Nervenarzt. 2017 Dec;88(12):1358-1366. doi: 10.1007/s00115-017-0447-3. Nervenarzt. 2017. PMID: 29101527 Review. German.
-
Spinal muscular atrophy: a time for screening.Curr Opin Pediatr. 2010 Dec;22(6):696-702. doi: 10.1097/MOP.0b013e32833f3046. Curr Opin Pediatr. 2010. PMID: 20829691 Review.
Cited by
-
Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study.Neuromuscul Disord. 2019 Nov;29(11):842-856. doi: 10.1016/j.nmd.2019.09.007. Epub 2019 Sep 12. Neuromuscul Disord. 2019. PMID: 31704158 Free PMC article. Clinical Trial.
-
Mass spectrometry for metabolomics analysis: Applications in neonatal and cancer screening.Mass Spectrom Rev. 2024 Jul-Aug;43(4):683-712. doi: 10.1002/mas.21826. Epub 2022 Dec 16. Mass Spectrom Rev. 2024. PMID: 36524560 Free PMC article. Review.
-
Response to: Alfred Sandrock, Wildon Farwell. Letter to the Editor, Comparisons Between Separately Conducted Clinical Trials: Letter to the Editor Regarding Dabbous O, Maru B, Jansen JP, Lorenzi M, Cloutier M, Guérin A, et al. Adv Ther (2019) 36(5):1164-76. doi:10.1007/s12325-019-00923-8.Adv Ther. 2019 Nov;36(11):2982-2985. doi: 10.1007/s12325-019-01088-0. Epub 2019 Sep 11. Adv Ther. 2019. PMID: 31512141 Free PMC article. No abstract available.
-
Comprehensive profile and natural history of pediatric patients with spinal muscular atrophy: A large retrospective study from China.Front Neurol. 2022 Dec 20;13:1038012. doi: 10.3389/fneur.2022.1038012. eCollection 2022. Front Neurol. 2022. PMID: 36605788 Free PMC article.
-
Rapid Molecular Diagnosis of Genetically Inherited Neuromuscular Disorders Using Next-Generation Sequencing Technologies.J Clin Med. 2022 May 12;11(10):2750. doi: 10.3390/jcm11102750. J Clin Med. 2022. PMID: 35628876 Free PMC article.
References
-
- Zerres K, Rudnik-Schoneborn S. Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Archives of neurology. 1995;52(5):518–23. - PubMed
-
- Munsat T, Davies K. Spinal muscular atrophy. 32nd ENMC International Workshop. Naarden, The Netherlands, 10–12 March 1995. Neuromuscular disorders : NMD. 1996;6(2):125–7. - PubMed
-
- Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nature genetics. 1997;16(3):265–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
