

GM2 Activator Deficiency Caused by a Homozygous Exon 2 Deletion in GM2A
- PMID: 28540636
- PMCID: PMC5874204
- DOI: 10.1007/8904_2017_31
GM2 Activator Deficiency Caused by a Homozygous Exon 2 Deletion in GM2A
Abstract
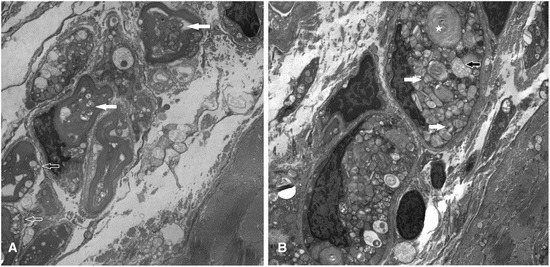
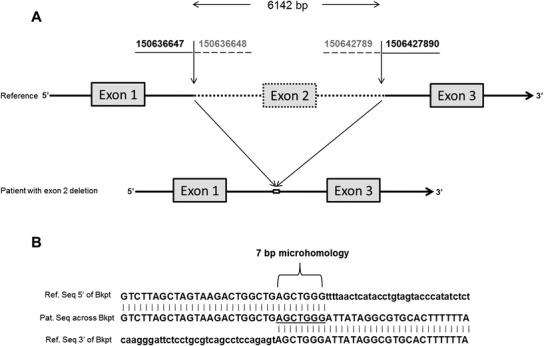
GM2 activator (GM2A) deficiency (OMIM 613109) is a rare lysosomal storage disorder, with onset typically in infancy or early childhood. Clinically, it is almost indistinguishable from Tay-Sachs disease (OMIM 272800) or Sandhoff disease (OMIM 268800); however, traditionally available biochemical screening tests will most likely reveal normal results. We report a 2-year-old male with initially normal development until the age of 9 months, when he presented with developmental delay and regression. Workup at that time was unrevealing; at 15 months, he had abnormal brain MRI findings and a cherry red spot on ophthalmological examination. Family history and all laboratory studies were uninformative. The combination of a cherry red spot and developmental regression was strongly suggestive of a lysosomal storage disorder. Sequence analysis of GM2A did not reveal any pathogenic variants; however, exon 2 of GM2A could not be amplified by PCR, raising suspicion for a large, homozygous deletion. Subsequent copy number analysis confirmed a homozygous deletion of exon 2 in GM2A. This is the first reported case of GM2A deficiency being caused by a whole exon deletion. We describe previously unreported electron microscopy findings in this disease, thus expanding the clinical and variant spectrum for GM2 activator deficiency. These findings demonstrate the increased degree of suspicion required for diagnosis of this rare disorder. Brief Summary: This case of GM2 activator deficiency was caused by a homozygous deletion in GM2A, demonstrating the need to include exon level copy number analysis in any workup to fully exclude this disorder.
Keywords: Cherry red spot; Copy number analysis; Electron microscopy; GM2 activator deficiency; GM2A; Lysosomal storage disorder.
Conflict of interest statement
Patricia Hall is employed by Emory University and is a laboratory director at EGL Genetic Diagnostics, LLC, a clinical genetics laboratory which performs testing described in this paper.
John Alexander is employed by Emory University and is a laboratory director at EGL Genetic Diagnostics, LLC, a clinical genetics laboratory which performs testing described in this paper.
Arun Ankala is employed by Emory University and is a laboratory director at EGL Genetic Diagnostics, LLC, a clinical genetics laboratory which performs testing described in this paper.
Regina Laine, Hart Lidov, Lisa Teot, and Irina Anselm declare that they have no conflict of interest.
Figures
Similar articles
-
Two patients from Turkey with a novel variant in the GM2A gene and review of the literature.J Pediatr Endocrinol Metab. 2021 Apr 6;34(6):805-812. doi: 10.1515/jpem-2020-0655. Print 2021 Jun 25. J Pediatr Endocrinol Metab. 2021. PMID: 33819415 Review.
-
GM2 gangliosidosis AB variant: novel mutation from India - a case report with a review.BMC Pediatr. 2016 Jul 11;16:88. doi: 10.1186/s12887-016-0626-6. BMC Pediatr. 2016. PMID: 27402091 Free PMC article. Review.
-
Combined replacement effects of human modified β-hexosaminidase B and GM2 activator protein on GM2 gangliosidoses fibroblasts.Biochem Biophys Rep. 2016 Jun 8;7:157-163. doi: 10.1016/j.bbrep.2016.04.012. eCollection 2016 Sep. Biochem Biophys Rep. 2016. PMID: 28955902 Free PMC article.
-
Atypical juvenile presentation of GM2 gangliosidosis AB in a patient compound-heterozygote for c.259G > T and c.164C > T mutations in the GM2A gene.Mol Genet Metab Rep. 2017 Apr 7;11:24-29. doi: 10.1016/j.ymgmr.2017.01.017. eCollection 2017 Jun. Mol Genet Metab Rep. 2017. PMID: 28417072 Free PMC article.
-
Structure of the GM2A gene: identification of an exon 2 nonsense mutation and a naturally occurring transcript with an in-frame deletion of exon 2.Am J Hum Genet. 1999 Jul;65(1):77-87. doi: 10.1086/302463. Am J Hum Genet. 1999. PMID: 10364519 Free PMC article.
Cited by
-
Multimodal optical imaging and genetic features of AB variant GM2 gangliosidosis: a case report.Front Pediatr. 2023 May 5;11:1147836. doi: 10.3389/fped.2023.1147836. eCollection 2023. Front Pediatr. 2023. PMID: 37215589 Free PMC article.
References
-
- Hall P, Minnich S, Teigen C, Raymond K. Diagnosing lysosomal storage disorders: the GM2 gangliosidoses. Curr Protoc Hum Genet. 2014;83:17.16.11–17.16.18. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
