

Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans
- PMID: 28346228
- PMCID: PMC5409795
- DOI: 10.1172/JCI91913
Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans
Abstract
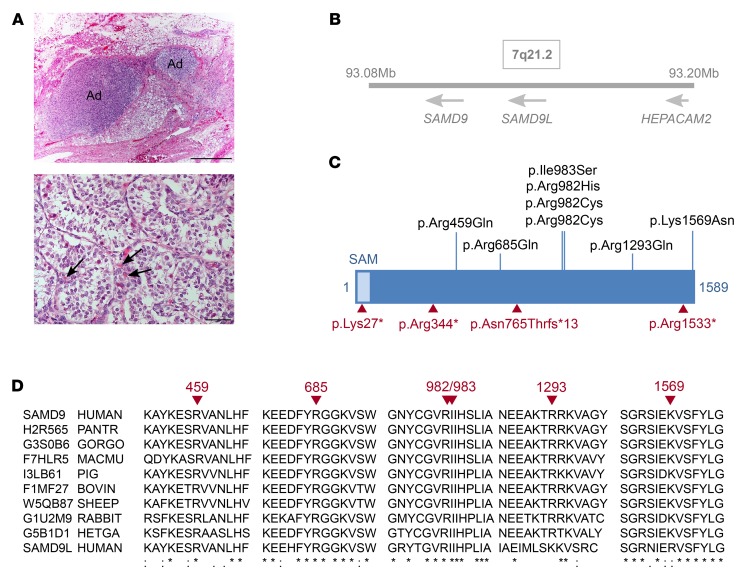
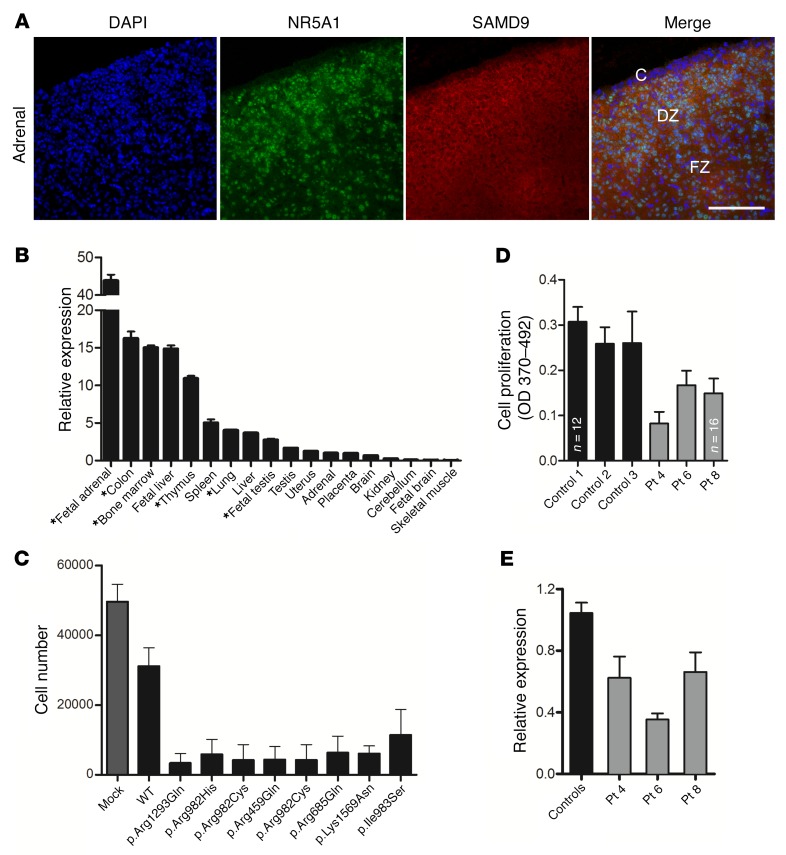
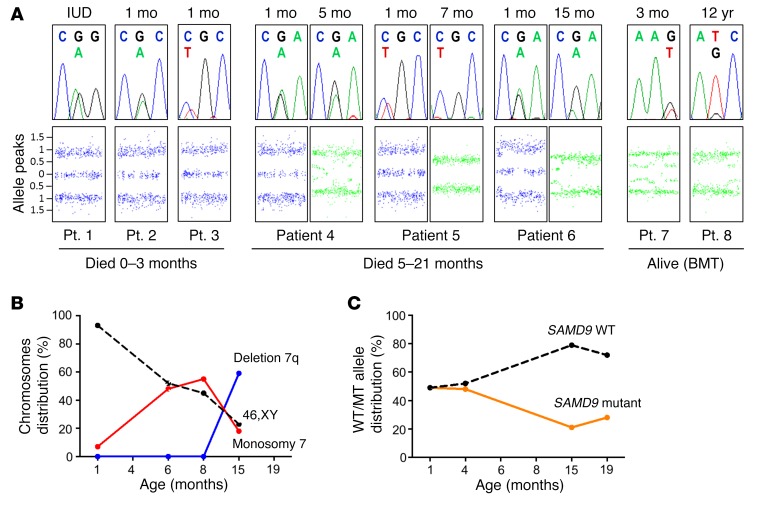
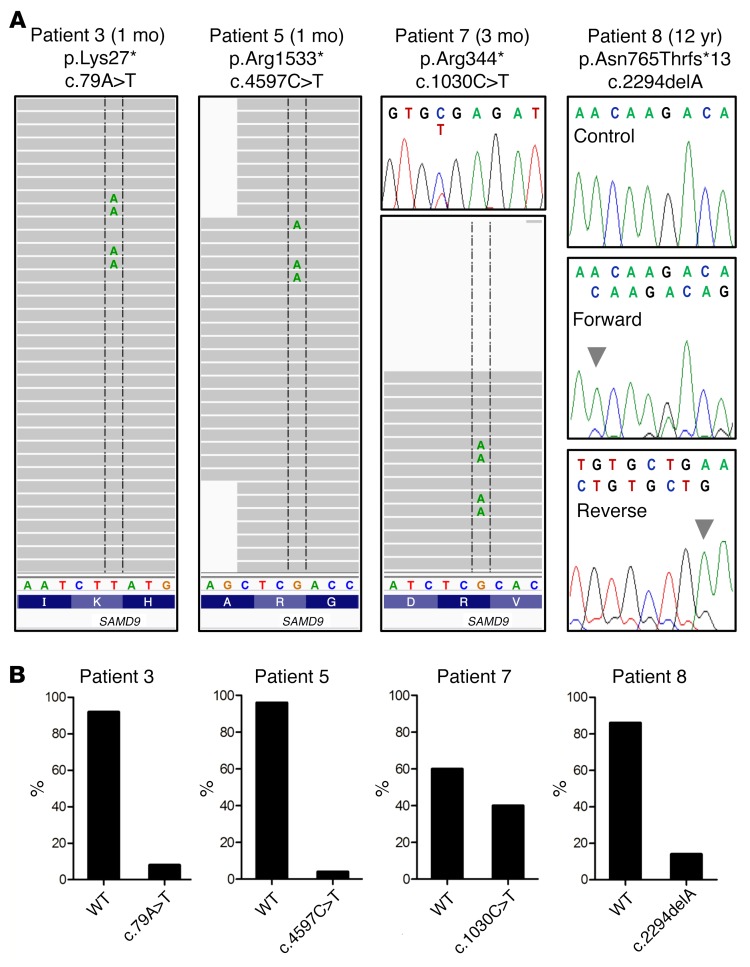
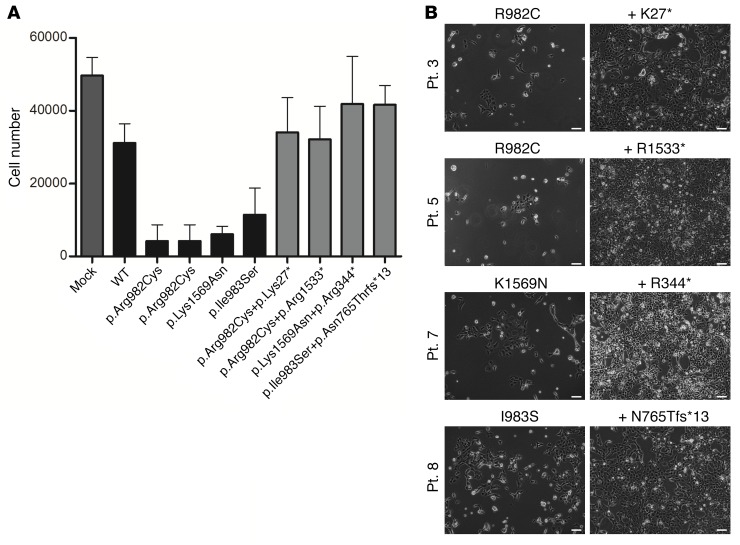
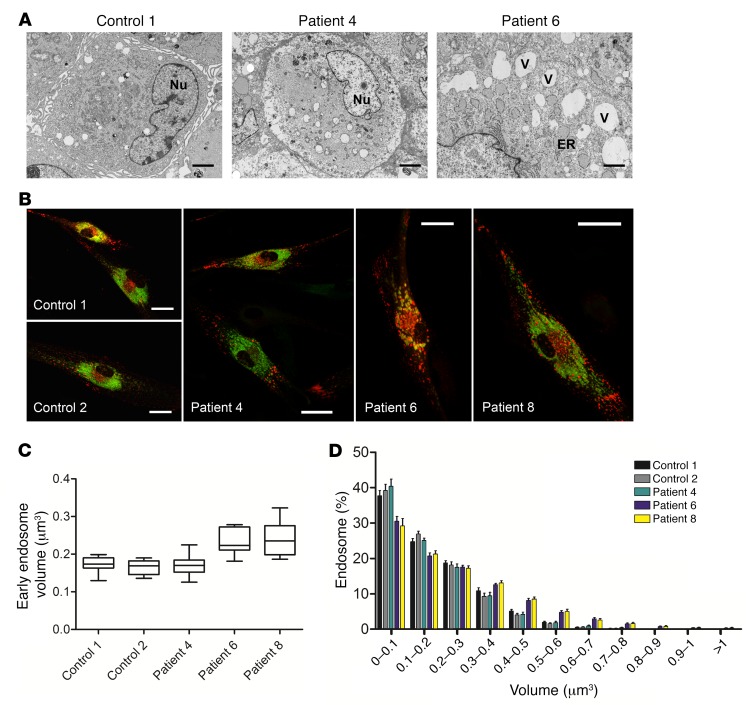
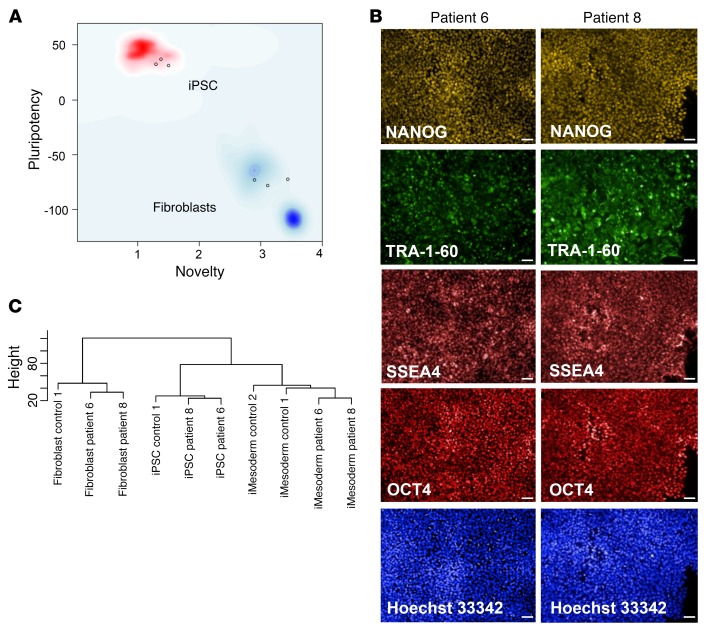
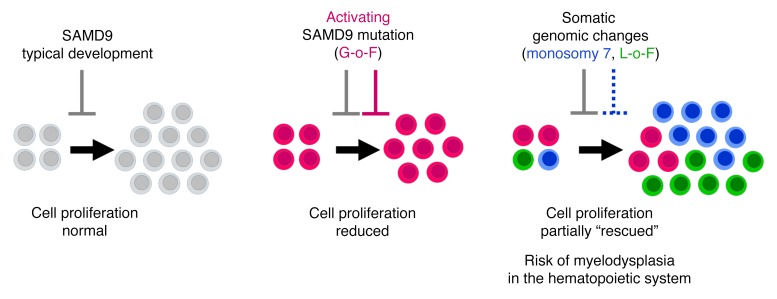
It is well established that somatic genomic changes can influence phenotypes in cancer, but the role of adaptive changes in developmental disorders is less well understood. Here we have used next-generation sequencing approaches to identify de novo heterozygous mutations in sterile α motif domain-containing protein 9 (SAMD9, located on chromosome 7q21.2) in 8 children with a multisystem disorder termed MIRAGE syndrome that is characterized by intrauterine growth restriction (IUGR) with gonadal, adrenal, and bone marrow failure, predisposition to infections, and high mortality. These mutations result in gain of function of the growth repressor product SAMD9. Progressive loss of mutated SAMD9 through the development of monosomy 7 (-7), deletions of 7q (7q-), and secondary somatic loss-of-function (nonsense and frameshift) mutations in SAMD9 rescued the growth-restricting effects of mutant SAMD9 proteins in bone marrow and was associated with increased length of survival. However, 2 patients with -7 and 7q- developed myelodysplastic syndrome, most likely due to haploinsufficiency of related 7q21.2 genes. Taken together, these findings provide strong evidence that progressive somatic changes can occur in specific tissues and can subsequently modify disease phenotype and influence survival. Such tissue-specific adaptability may be a more common mechanism modifying the expression of human genetic conditions than is currently recognized.
Conflict of interest statement
Figures
Similar articles
-
Emerging phenotypes linked to variants in SAMD9 and MIRAGE syndrome.Front Endocrinol (Lausanne). 2022 Aug 18;13:953707. doi: 10.3389/fendo.2022.953707. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36060959 Free PMC article.
-
Somatic mosaic monosomy 7 and UPD7q in a child with MIRAGE syndrome caused by a novel SAMD9 mutation.Pediatr Blood Cancer. 2019 Apr;66(4):e27589. doi: 10.1002/pbc.27589. Epub 2018 Dec 19. Pediatr Blood Cancer. 2019. PMID: 30565860
-
SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7.Nat Genet. 2016 Jul;48(7):792-7. doi: 10.1038/ng.3569. Epub 2016 May 16. Nat Genet. 2016. PMID: 27182967
-
Of gains and losses: SAMD9/SAMD9L and monosomy 7 in myelodysplastic syndrome.Exp Hematol. 2024 Jun;134:104217. doi: 10.1016/j.exphem.2024.104217. Epub 2024 Apr 20. Exp Hematol. 2024. PMID: 38649131 Review.
-
SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies.Leukemia. 2018 May;32(5):1106-1115. doi: 10.1038/s41375-018-0074-4. Epub 2018 Feb 25. Leukemia. 2018. PMID: 29535429 Free PMC article. Review.
Cited by
-
MIRAGE syndrome with recurrent pneumonia probably associated with gastroesophageal reflux and achalasia: A case report.Clin Pediatr Endocrinol. 2019;28(4):147-153. doi: 10.1297/cpe.28.147. Epub 2019 Oct 19. Clin Pediatr Endocrinol. 2019. PMID: 31666768 Free PMC article.
-
Prognostic mutation constellations in acute myeloid leukaemia and myelodysplastic syndrome.Curr Opin Hematol. 2021 Mar 1;28(2):101-109. doi: 10.1097/MOH.0000000000000629. Curr Opin Hematol. 2021. PMID: 33427759 Free PMC article. Review.
-
Clonal evolution in inherited marrow failure syndromes predicts disease progression.Hematology Am Soc Hematol Educ Program. 2023 Dec 8;2023(1):125-134. doi: 10.1182/hematology.2023000469. Hematology Am Soc Hematol Educ Program. 2023. PMID: 38066914 Free PMC article. Review.
-
Mutations in both SAMD9 and SLC19A2 genes caused complex phenotypes characterized by recurrent infection, dysphagia and profound deafness - a case report for dual diagnosis.BMC Pediatr. 2019 Oct 21;19(1):364. doi: 10.1186/s12887-019-1733-y. BMC Pediatr. 2019. PMID: 31638924 Free PMC article.
-
Primary adrenal insufficiency: New genetic causes and their long-term consequences.Clin Endocrinol (Oxf). 2020 Jan;92(1):11-20. doi: 10.1111/cen.14109. Epub 2019 Oct 30. Clin Endocrinol (Oxf). 2020. PMID: 31610036 Free PMC article. Review.
References
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
