

Prevalence and architecture of de novo mutations in developmental disorders
- PMID: 28135719
- PMCID: PMC6016744
- DOI: 10.1038/nature21062
Prevalence and architecture of de novo mutations in developmental disorders
Abstract
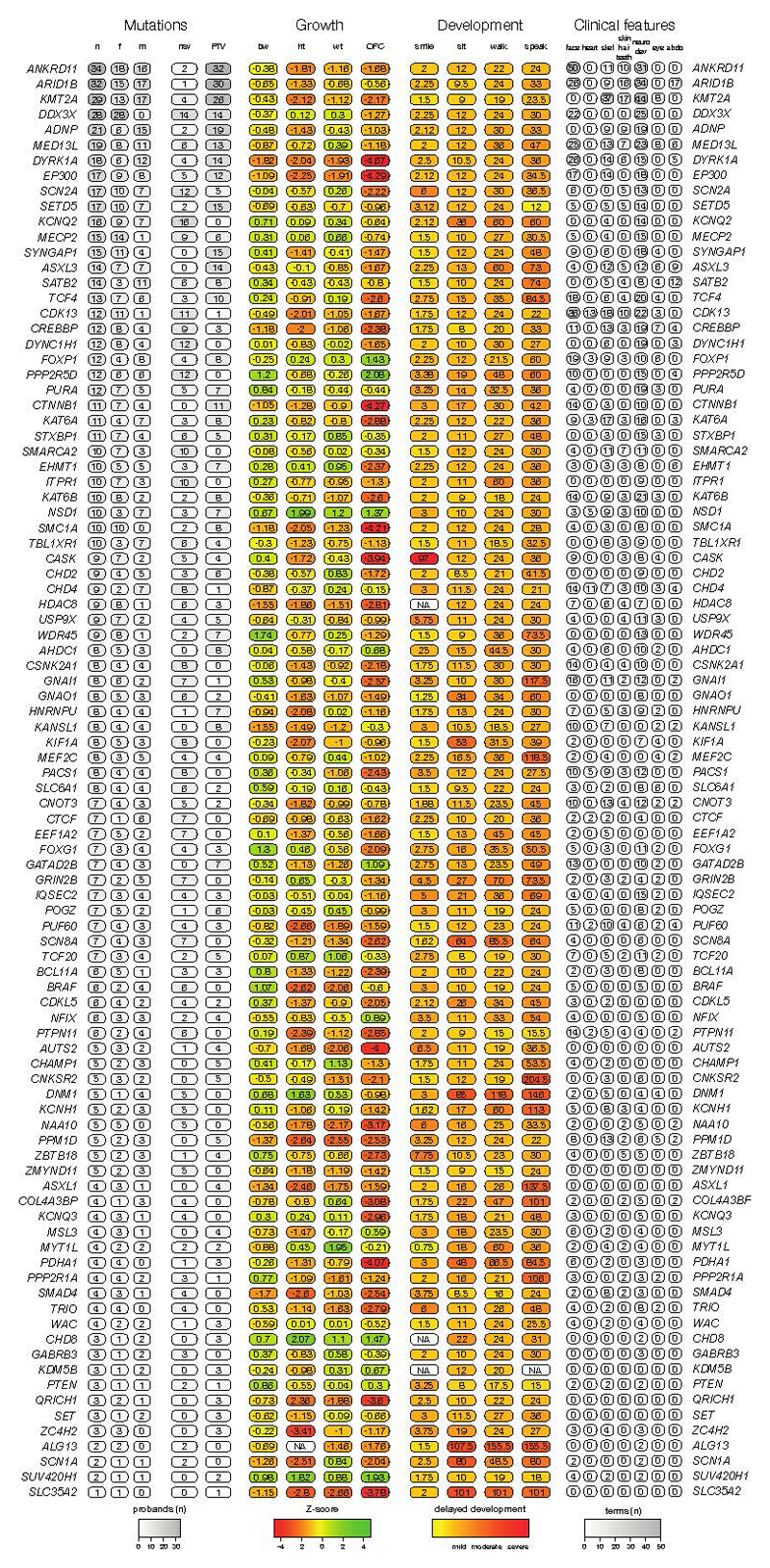
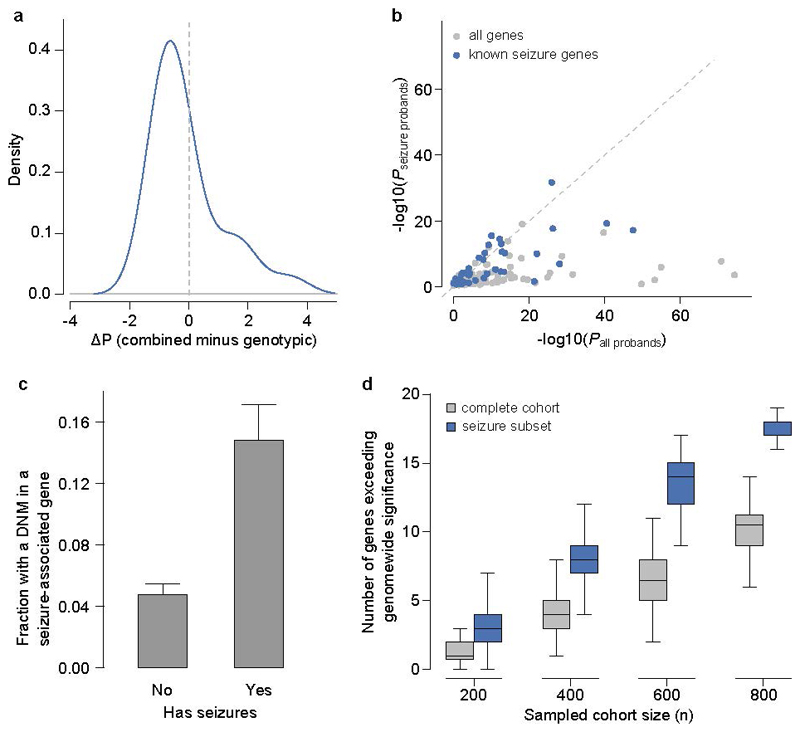
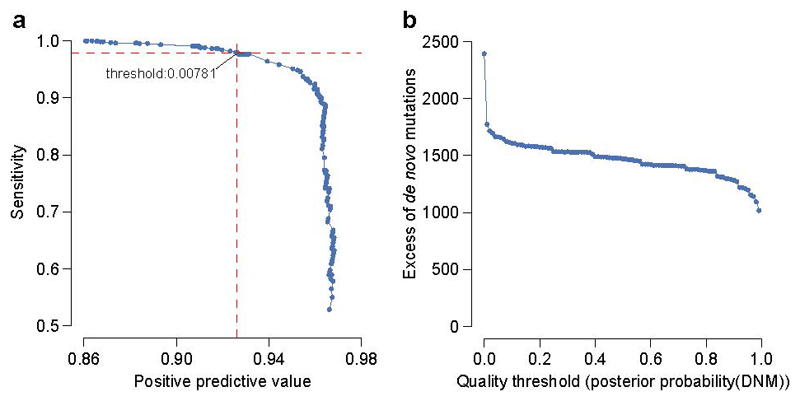
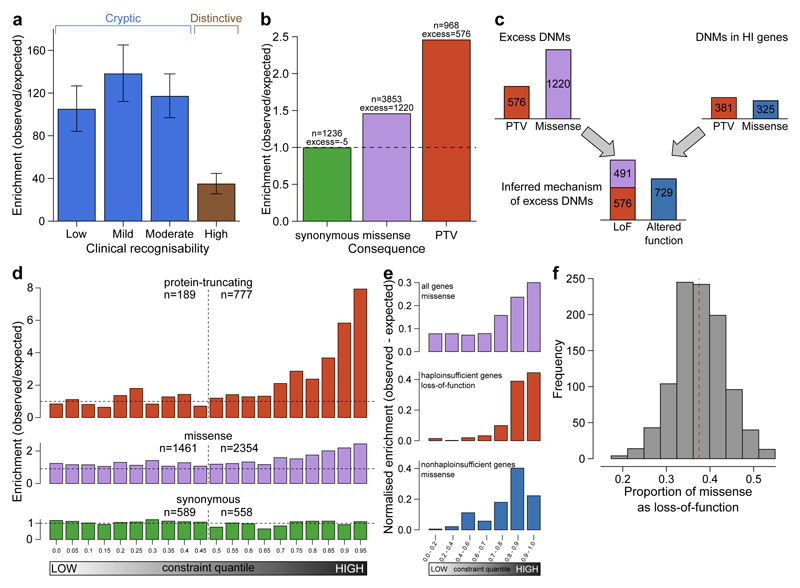
The genomes of individuals with severe, undiagnosed developmental disorders are enriched in damaging de novo mutations (DNMs) in developmentally important genes. Here we have sequenced the exomes of 4,293 families containing individuals with developmental disorders, and meta-analysed these data with data from another 3,287 individuals with similar disorders. We show that the most important factors influencing the diagnostic yield of DNMs are the sex of the affected individual, the relatedness of their parents, whether close relatives are affected and the parental ages. We identified 94 genes enriched in damaging DNMs, including 14 that previously lacked compelling evidence of involvement in developmental disorders. We have also characterized the phenotypic diversity among these disorders. We estimate that 42% of our cohort carry pathogenic DNMs in coding sequences; approximately half of these DNMs disrupt gene function and the remainder result in altered protein function. We estimate that developmental disorders caused by DNMs have an average prevalence of 1 in 213 to 1 in 448 births, depending on parental age. Given current global demographics, this equates to almost 400,000 children born per year.
Figures









Similar articles
-
Large-scale discovery of novel genetic causes of developmental disorders.Nature. 2015 Mar 12;519(7542):223-8. doi: 10.1038/nature14135. Epub 2014 Dec 24. Nature. 2015. PMID: 25533962 Free PMC article.
-
De novo damaging variants associated with congenital heart diseases contribute to the connectome.Sci Rep. 2020 Apr 27;10(1):7046. doi: 10.1038/s41598-020-63928-2. Sci Rep. 2020. PMID: 32341405 Free PMC article.
-
Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing.Nat Genet. 2016 Sep;48(9):1060-5. doi: 10.1038/ng.3627. Epub 2016 Aug 1. Nat Genet. 2016. PMID: 27479907 Free PMC article.
-
Developmental delay and failure to thrive associated with a loss-of-function variant in WHSC1 (NSD2).Am J Med Genet A. 2018 Dec;176(12):2798-2802. doi: 10.1002/ajmg.a.40498. Epub 2018 Oct 22. Am J Med Genet A. 2018. PMID: 30345613 Review.
-
Novel NR2F1 variants likely disrupt DNA binding: molecular modeling in two cases, review of published cases, genotype-phenotype correlation, and phenotypic expansion of the Bosch-Boonstra-Schaaf optic atrophy syndrome.Cold Spring Harb Mol Case Stud. 2017 Nov 21;3(6):a002162. doi: 10.1101/mcs.a002162. Print 2017 Nov. Cold Spring Harb Mol Case Stud. 2017. PMID: 28963436 Free PMC article. Review.
Cited by
-
Spatiotemporal 7q11.23 protein network analysis implicates the role of DNA repair pathway during human brain development.Sci Rep. 2021 Apr 15;11(1):8246. doi: 10.1038/s41598-021-87632-x. Sci Rep. 2021. PMID: 33859276 Free PMC article.
-
The chromatin remodeling enzyme Chd4 regulates genome architecture in the mouse brain.Nat Commun. 2020 Jul 9;11(1):3419. doi: 10.1038/s41467-020-17065-z. Nat Commun. 2020. PMID: 32647123 Free PMC article.
-
The MED13L haploinsufficiency syndrome associated with de novo nonsense variant (P.GLN1981*).J Mother Child. 2021 Apr 30;24(3):32-36. doi: 10.34763/jmotherandchild.20202403.2021.d-20-00003. J Mother Child. 2021. PMID: 33930262 Free PMC article.
-
Trio-based whole exome sequencing in patients with suspected sporadic inborn errors of immunity: A retrospective cohort study.Elife. 2022 Oct 17;11:e78469. doi: 10.7554/eLife.78469. Elife. 2022. PMID: 36250618 Free PMC article.
-
Evolutionarily new genes in humans with disease phenotypes reveal functional enrichment patterns shaped by adaptive innovation and sexual selection.Res Sq [Preprint]. 2023 Nov 21:rs.3.rs-3632644. doi: 10.21203/rs.3.rs-3632644/v1. Res Sq. 2023. Update in: Genome Res. 2025 Mar 4. doi: 10.1101/gr.279498.124. PMID: 38045389 Free PMC article. Updated. Preprint.
References
-
- Sheridan E, et al. Risk factors for congenital anomaly in a multiethnic birth cohort: an analysis of the Born in Bradford study. Lancet. 2013;382:1350–9. - PubMed
-
- Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet. 2010;11:161–87. - PubMed
-
- De Ligt J, et al. Diagnostic exome sequencing in persons with severe intellectual disability. The New England Journal of Medicine. 2012;367:1921–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
