

Sphingosine 1-phosphate lyase deficiency causes Charcot-Marie-Tooth neuropathy
- PMID: 28077491
- PMCID: PMC5304460
- DOI: 10.1212/WNL.0000000000003595
Sphingosine 1-phosphate lyase deficiency causes Charcot-Marie-Tooth neuropathy
Abstract
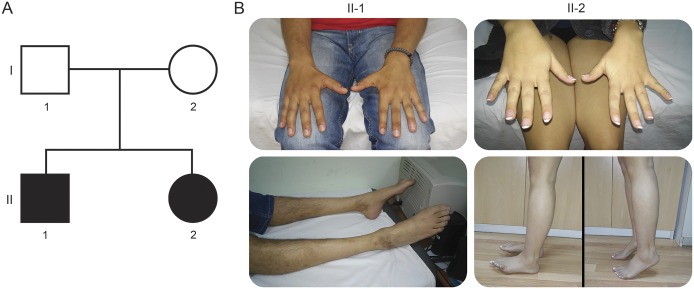
Objective: To identify the unknown genetic cause in a nuclear family with an axonal form of peripheral neuropathy and atypical disease course.
Methods: Detailed neurologic, electrophysiologic, and neuropathologic examinations of the patients were performed. Whole exome sequencing of both affected individuals was done. The effect of the identified sequence variations was investigated at cDNA and protein level in patient-derived lymphoblasts. The plasma sphingoid base profile was analyzed. Functional consequences of neuron-specific downregulation of the gene were studied in Drosophila.
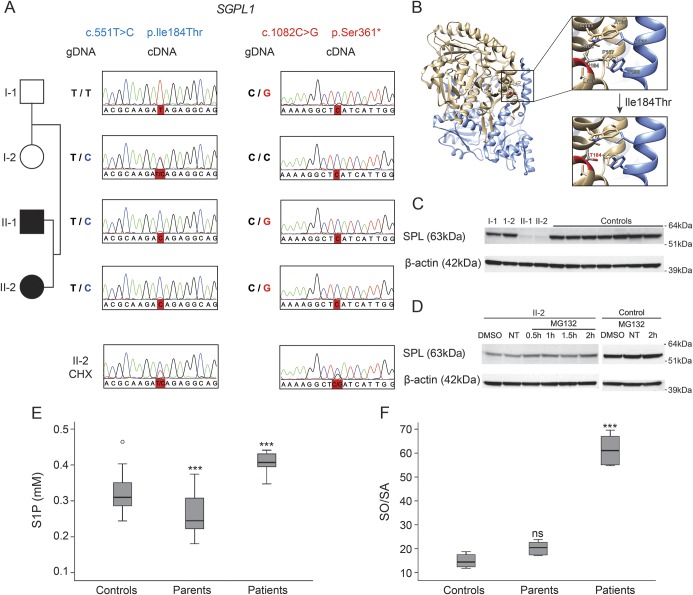
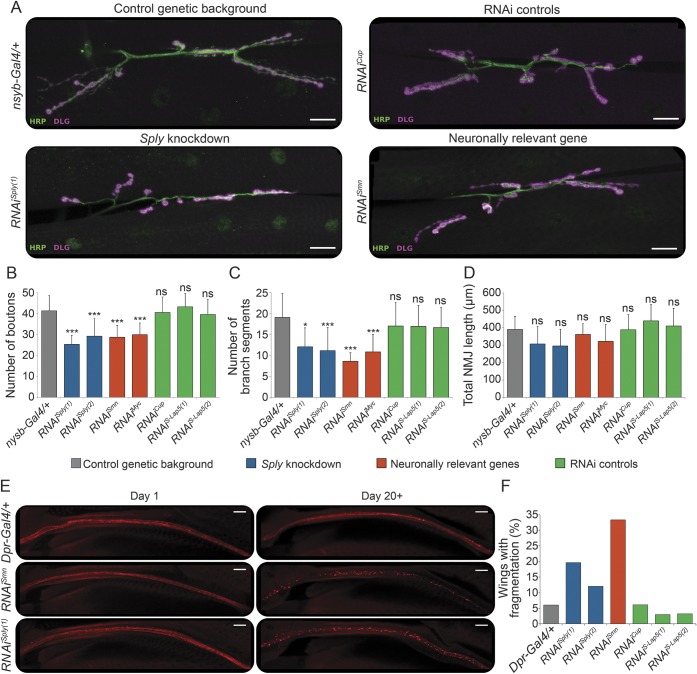
Results: Both patients present an atypical form of axonal peripheral neuropathy, characterized by acute or subacute onset and episodes of recurrent mononeuropathy. We identified compound heterozygous mutations cosegregating with disease and absent in controls in the SGPL1 gene, encoding sphingosine 1-phosphate lyase (SPL). The p.Ser361* mutation triggers nonsense-mediated mRNA decay. The missense p.Ile184Thr mutation causes partial protein degradation. The plasma levels of sphingosine 1-phosphate and sphingosine/sphinganine ratio were increased in the patients. Neuron-specific downregulation of the Drosophila orthologue impaired the morphology of the neuromuscular junction and caused progressive degeneration of the chemosensory neurons innervating the wing margin bristles.
Conclusions: We suggest SPL deficiency as a cause of a distinct form of Charcot-Marie-Tooth disease in humans, thus extending the currently recognized clinical and genetic spectrum of inherited peripheral neuropathies. Our data emphasize the importance of sphingolipid metabolism for neuronal function.
© 2017 American Academy of Neurology.
Figures
Similar articles
-
Genetic epidemiology of Charcot-Marie-Tooth disease.Acta Neurol Scand Suppl. 2012;(193):iv-22. doi: 10.1111/ane.12013. Acta Neurol Scand Suppl. 2012. PMID: 23106488
-
Sphingosine phosphate lyase insufficiency syndrome (SPLIS): A novel inborn error of sphingolipid metabolism.Adv Biol Regul. 2019 Jan;71:128-140. doi: 10.1016/j.jbior.2018.09.004. Epub 2018 Sep 25. Adv Biol Regul. 2019. PMID: 30274713 Free PMC article. Review.
-
Deficiency of the sphingosine-1-phosphate lyase SGPL1 is associated with congenital nephrotic syndrome and congenital adrenal calcifications.Hum Mutat. 2017 Apr;38(4):365-372. doi: 10.1002/humu.23192. Epub 2017 Mar 6. Hum Mutat. 2017. PMID: 28181337 Free PMC article.
-
Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency.J Clin Invest. 2017 Mar 1;127(3):912-928. doi: 10.1172/JCI89626. Epub 2017 Feb 6. J Clin Invest. 2017. PMID: 28165339 Free PMC article.
-
Genotype/Phenotype Interactions and First Steps Toward Targeted Therapy for Sphingosine Phosphate Lyase Insufficiency Syndrome.Cell Biochem Biophys. 2021 Sep;79(3):547-559. doi: 10.1007/s12013-021-01013-9. Epub 2021 Jun 16. Cell Biochem Biophys. 2021. PMID: 34133011 Review.
Cited by
-
Alternative splicing expands the clinical spectrum of NDUFS6-related mitochondrial disorders.Genet Med. 2024 Jun;26(6):101117. doi: 10.1016/j.gim.2024.101117. Epub 2024 Mar 6. Genet Med. 2024. PMID: 38459834 Free PMC article.
-
N-Acetylcysteine Alleviates Impaired Muscular Function Resulting from Sphingosine Phosphate Lyase Functional Deficiency-Induced Sphingoid Base and Ceramide Accumulation in Caenorhabditis elegans.Nutrients. 2024 May 26;16(11):1623. doi: 10.3390/nu16111623. Nutrients. 2024. PMID: 38892556 Free PMC article.
-
Sphingolipids in neurodegenerative diseases.Front Neurosci. 2023 Feb 16;17:1137893. doi: 10.3389/fnins.2023.1137893. eCollection 2023. Front Neurosci. 2023. PMID: 36875645 Free PMC article. Review.
-
Fifty years of lyase and a moment of truth: sphingosine phosphate lyase from discovery to disease.J Lipid Res. 2019 Mar;60(3):456-463. doi: 10.1194/jlr.S091181. Epub 2019 Jan 11. J Lipid Res. 2019. PMID: 30635364 Free PMC article. Review.
-
Transcriptional dysregulation by a nucleus-localized aminoacyl-tRNA synthetase associated with Charcot-Marie-Tooth neuropathy.Nat Commun. 2019 Nov 6;10(1):5045. doi: 10.1038/s41467-019-12909-9. Nat Commun. 2019. PMID: 31695036 Free PMC article.
References
-
- Reilly MM, Murphy SM, Laura M. Charcot-Marie-Tooth disease. J Peripher Nervous Syst 2011;16:1–14. - PubMed
-
- Ouvrier R. What can we learn from the history of Charcot-Marie-Tooth disease? Dev Med Child Neurol 2010;52:405–406. - PubMed
-
- Davis CJ, Bradley WG, Madrid R. The peroneal muscular atrophy syndrome: clinical, genetic, electrophysiological and nerve biopsy studies: I: clinical, genetic and electrophysiological findings and classification. J Genet Hum 1978;26:311–349. - PubMed
-
- Dubourg O, Azzedine H, Verny C, et al. . Autosomal-recessive forms of demyelinating Charcot-Marie-Tooth disease. Neuromolecular Med 2006;8:75–86. - PubMed
-
- Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol 2013;9:562–571. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases