

Childhood neurofibromatosis type 2 (NF2) and related disorders: from bench to bedside and biologically targeted therapies
- PMID: 27958595
- PMCID: PMC5225790
- DOI: 10.14639/0392-100X-1093
Childhood neurofibromatosis type 2 (NF2) and related disorders: from bench to bedside and biologically targeted therapies
Abstract
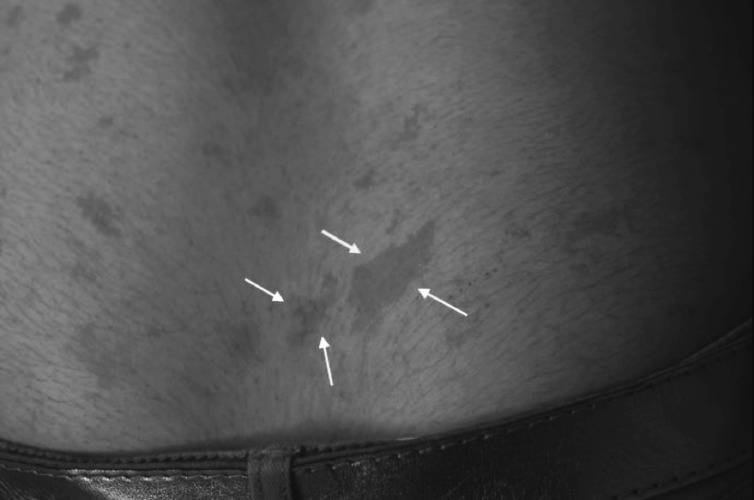
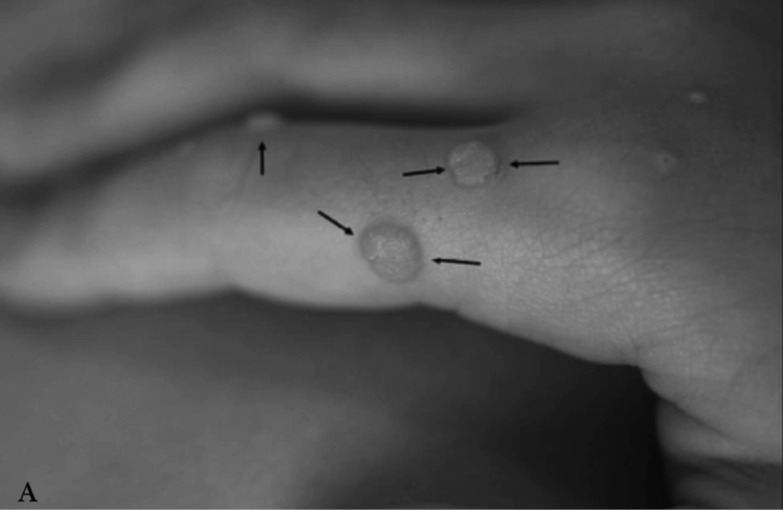
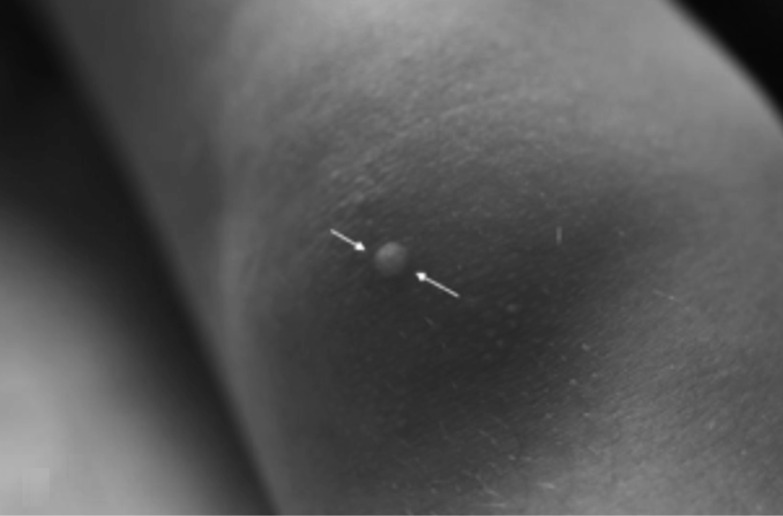
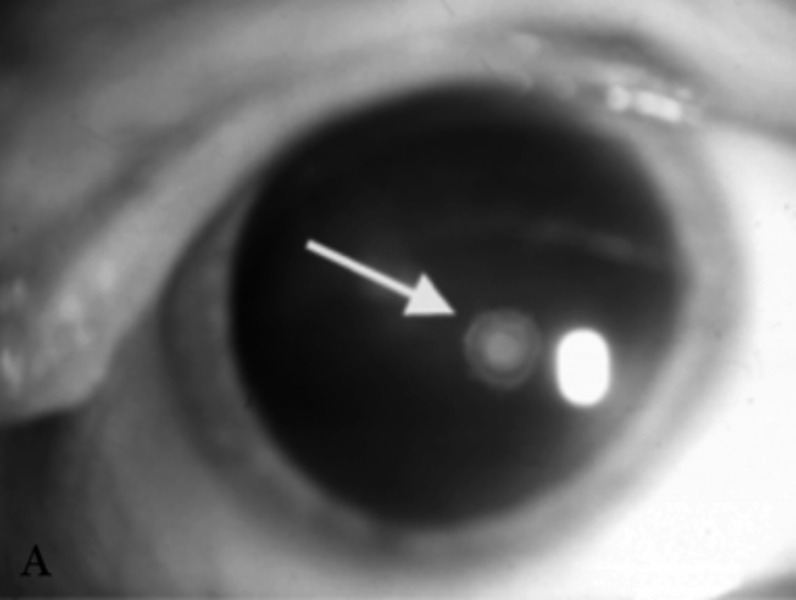
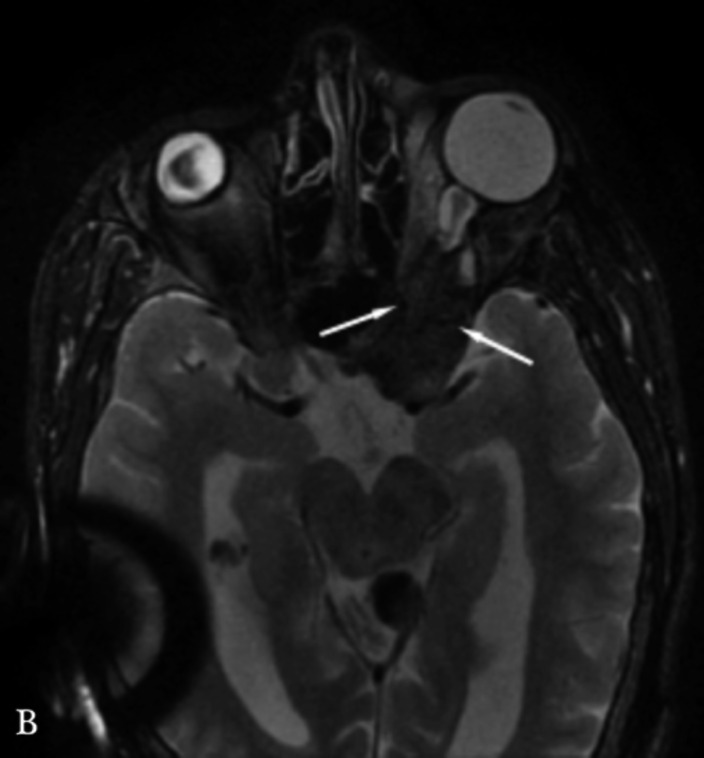
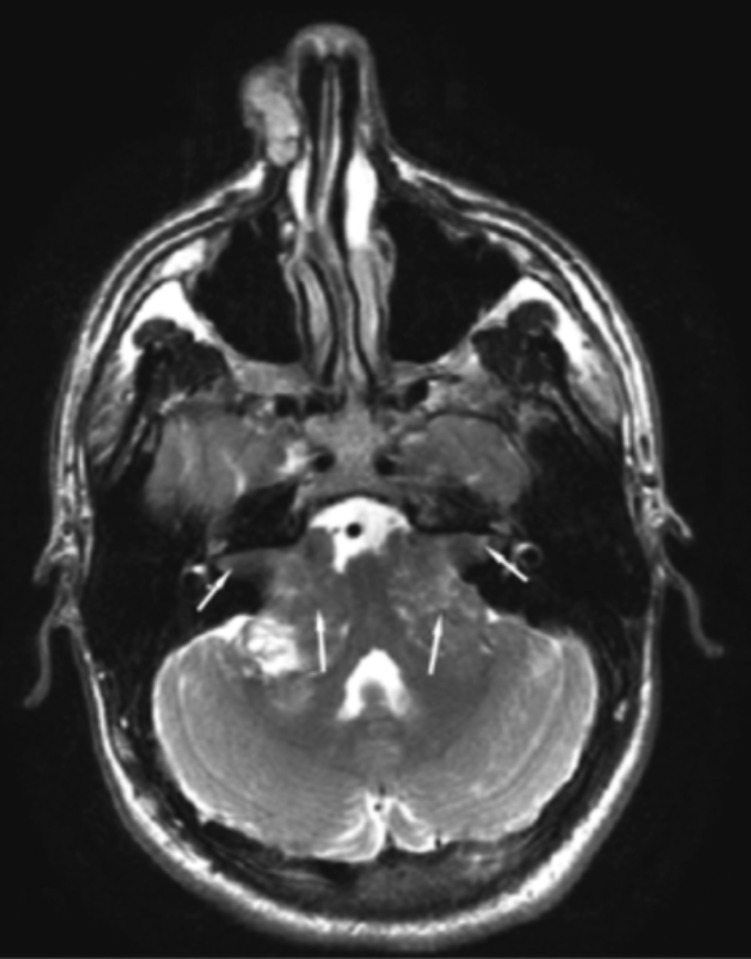
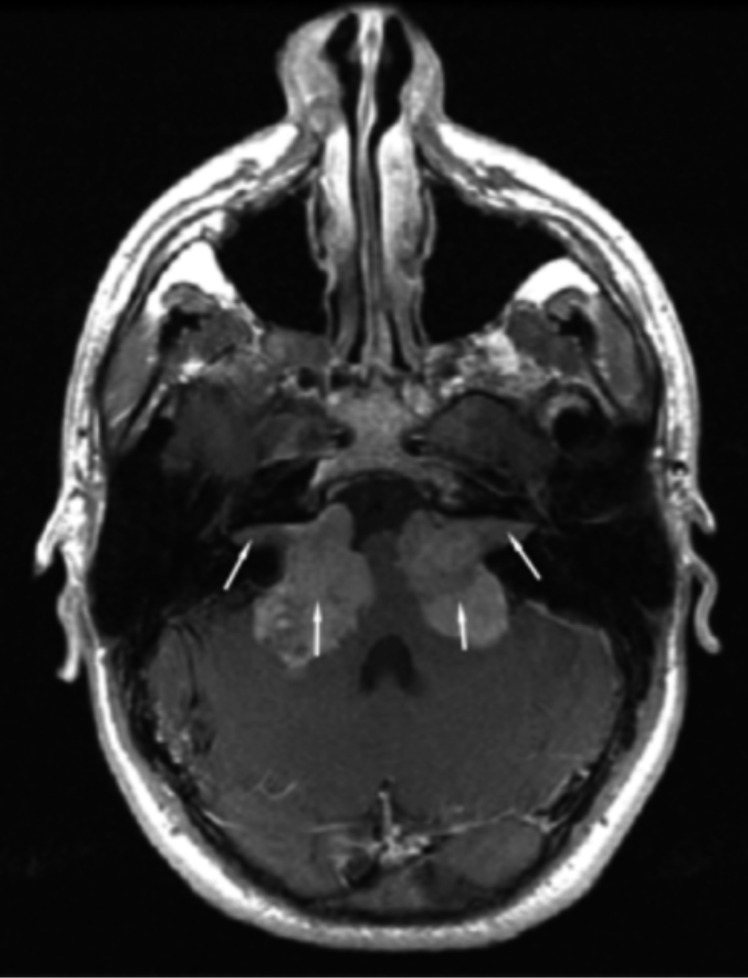
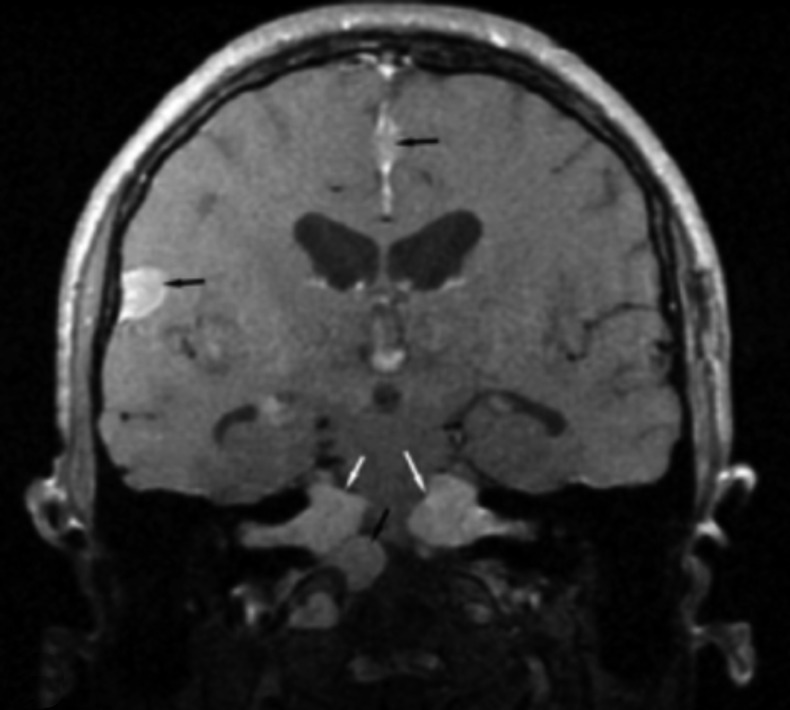
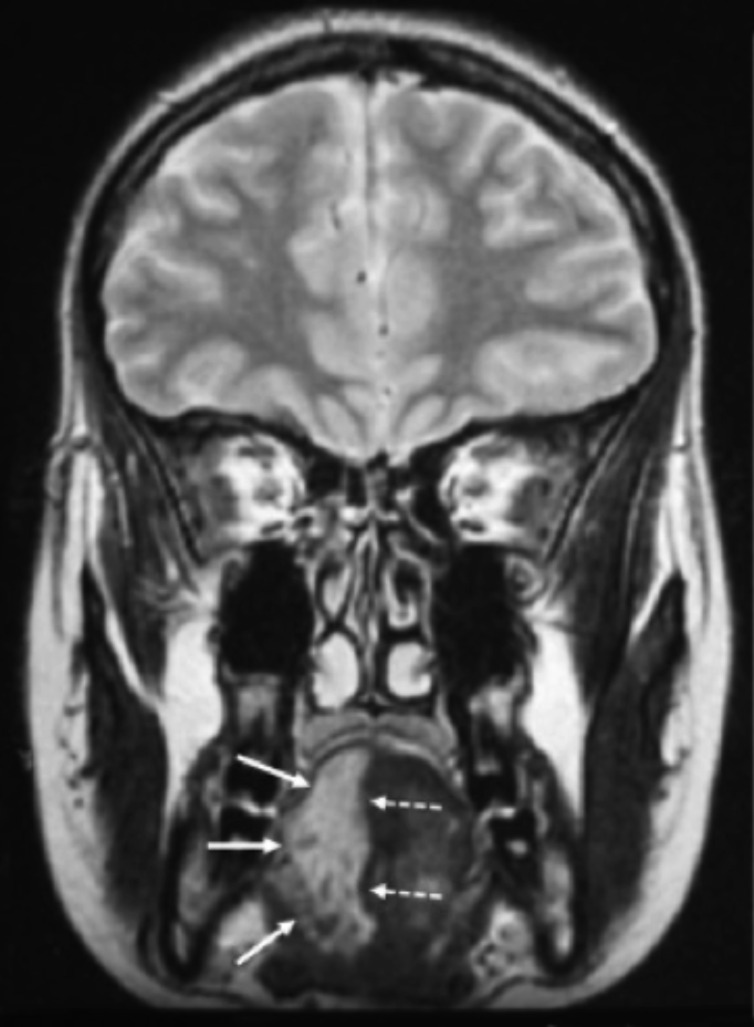
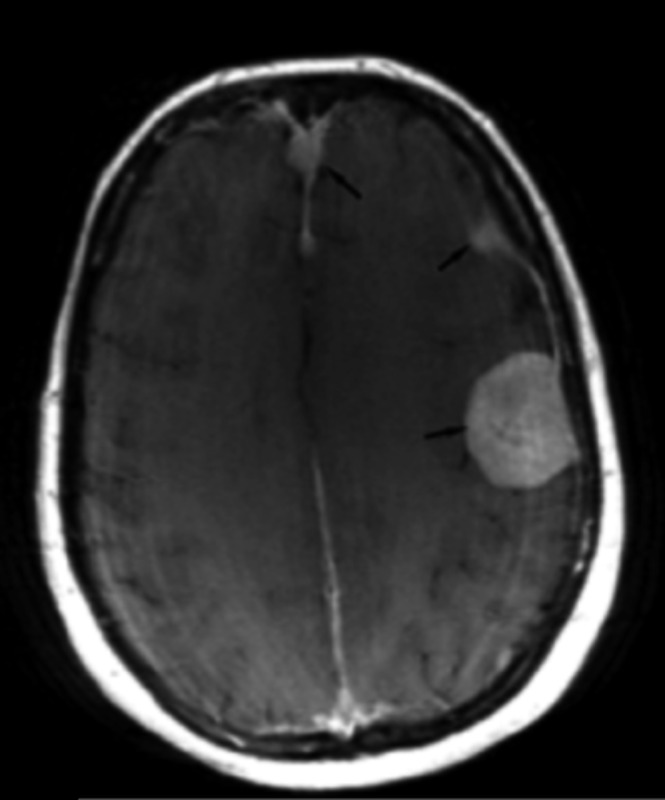
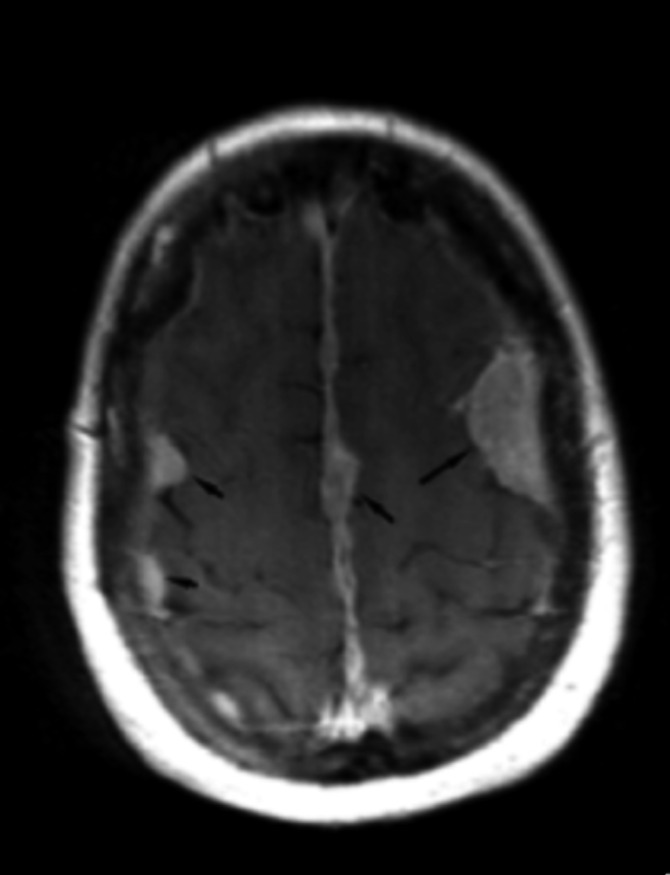
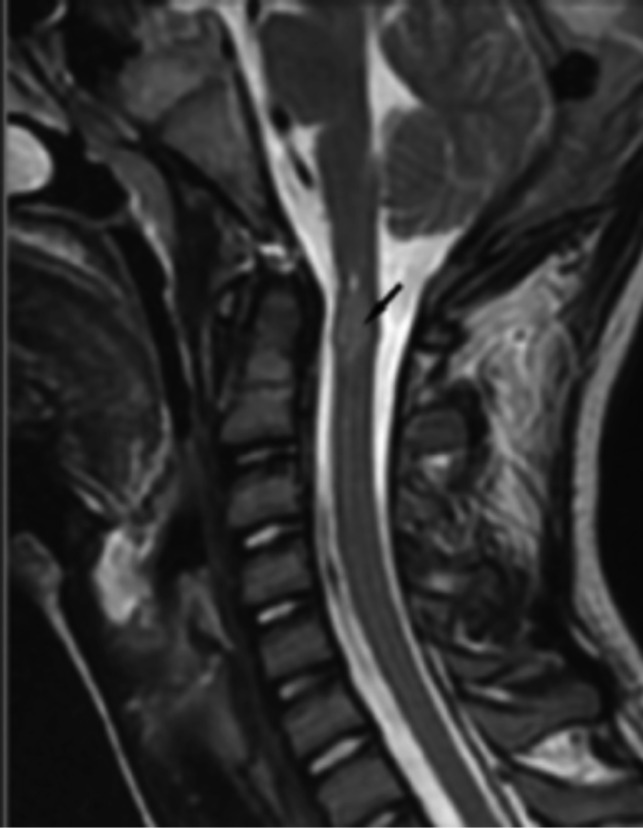
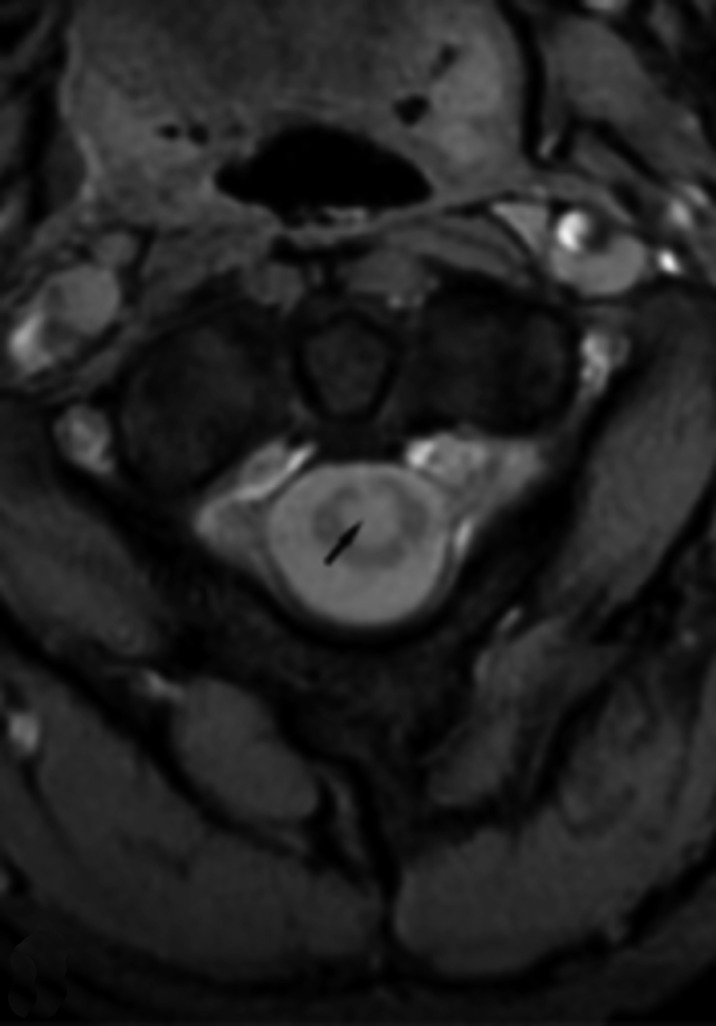
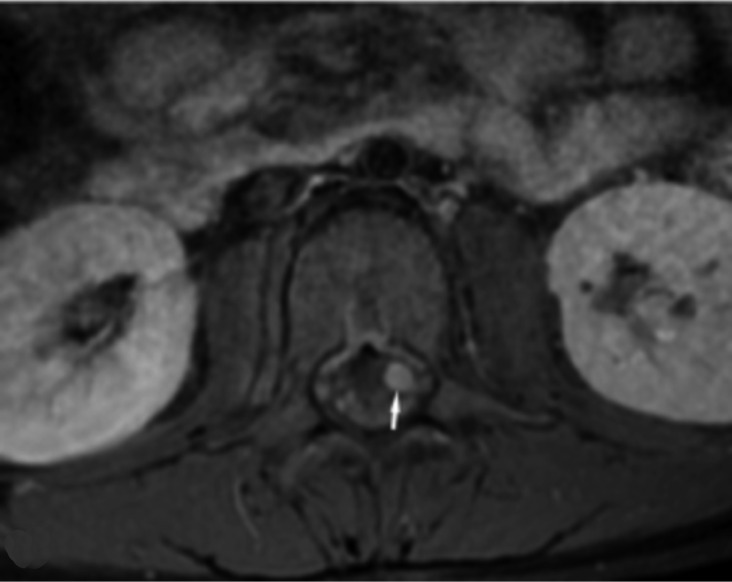
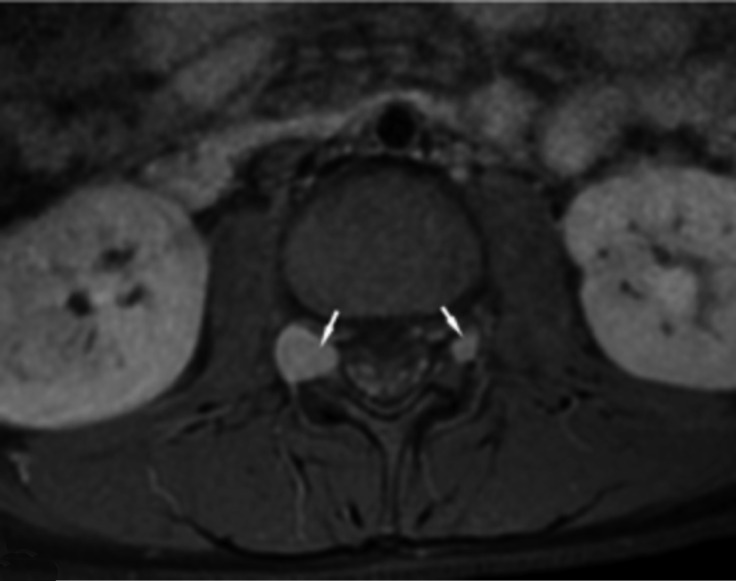
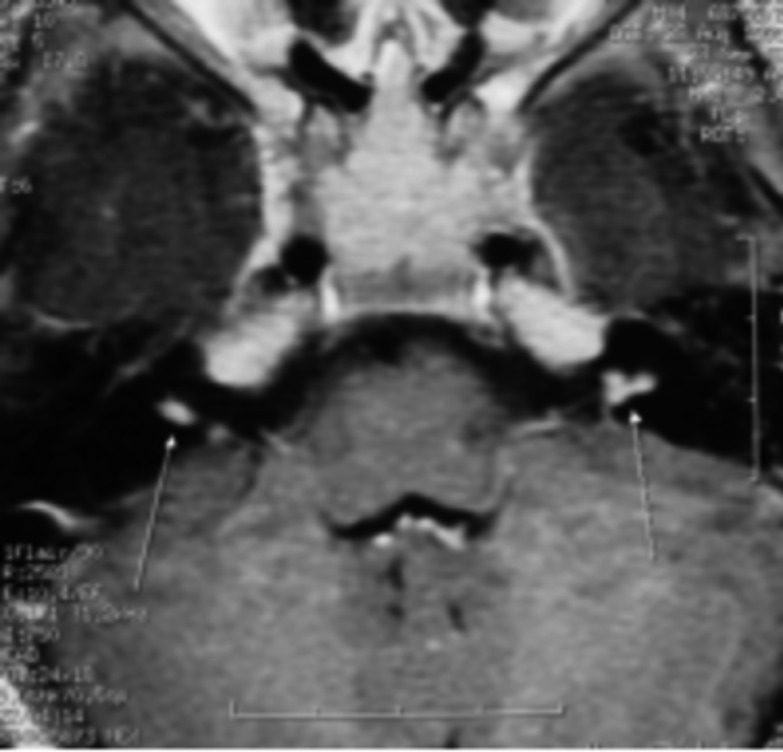
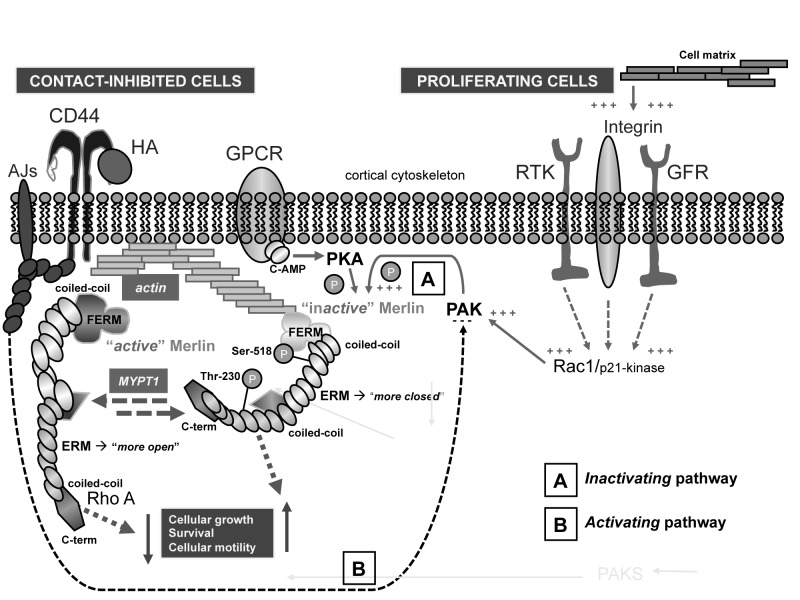
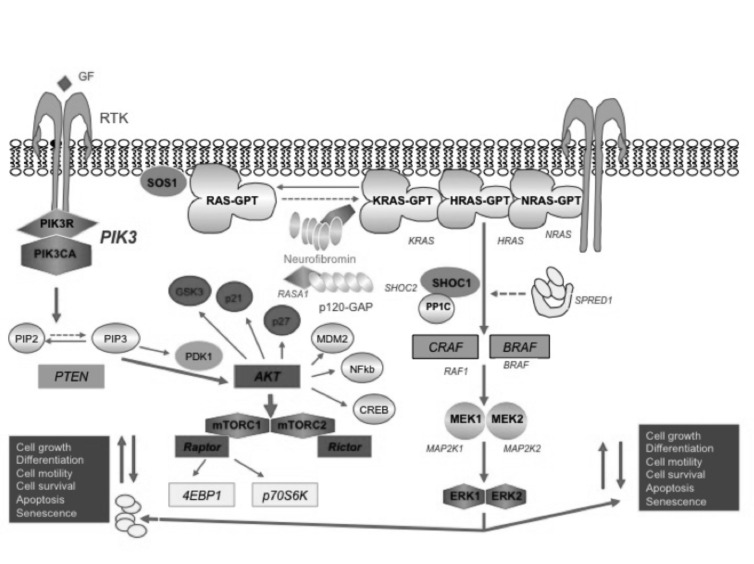
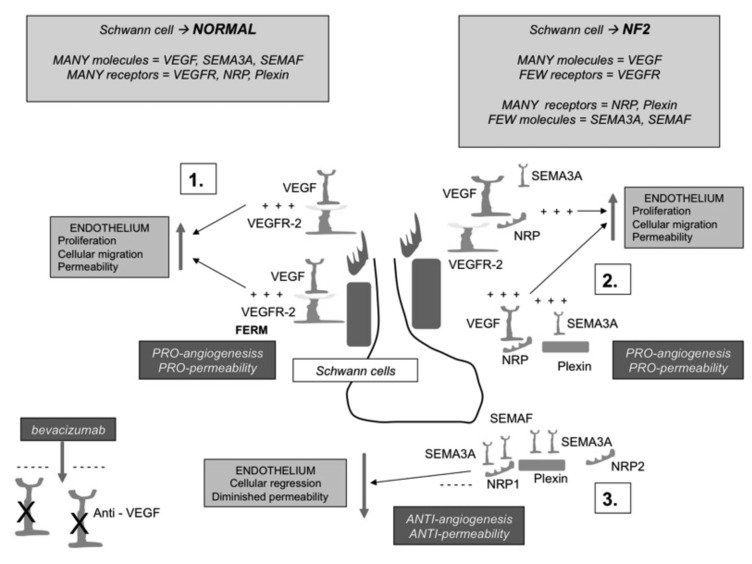
Neurofibromatosis type 2 [NF2; MIM # 101000] is an autosomal dominant disorder characterised by the occurrence of vestibular schwannomas (VSs), schwannomas of other cranial, spinal and cutaneous nerves, cranial and spinal meningiomas and/or other central nervous system (CNS) tumours (e.g., ependymomas, astrocytomas). Additional features include early onset cataracts, optic nerve sheath meningiomas, retinal hamartomas, dermal schwannomas (i.e., NF2-plaques), and (few) café-au-lait spots. Clinically, NF2 children fall into two main groups: (1) congenital NF2 - with bilateral VSs detected as early as the first days to months of life, which can be stable/asymptomatic for one-two decades and suddenly progress; and (2) severe pre-pubertal (Wishart type) NF2- with multiple (and rapidly progressive) CNS tumours other-than-VS, which usually present first, years before VSs [vs. the classical adult (Gardner type) NF2, with bilateral VSs presenting in young adulthood, sometimes as the only disease feature]. Some individuals can develop unilateral VS associated with ipsilateral meningiomas or multiple schwannomas localised to one part of the peripheral nervous system [i.e., mosaic NF2] or multiple non-VS, non-intradermal cranial, spinal and peripheral schwannomas (histologically proven) [schwannomatosis]. NF2 is caused by mutations in the NF2 gene at chromosome 22q12.1, which encodes for a protein called merlin or schwannomin, most similar to the exrin-readixin-moesin (ERM) proteins; mosaicNF2 is due to mosaic phenomena for the NF2 gene, whilst schwannomatosis is caused by coupled germ-line and mosaic mutations either in the SMARCB1 gene [SWNTS1; MIM # 162091] or the LZTR1 gene [SWNTS2; MIM # 615670] both falling within the 22q region and the NF2 gene. Data driven from in vitro and animal studies on the merlin pathway [e.g., post-translational and upstream/downstream regulation] allowed biologically targeted treatment strategies [e.g., Lapatinib, Erlotinib, Bevacizumab] aimed to multiple tumour shrinkage and/or regression and tumour arrest of progression with functional improvement.
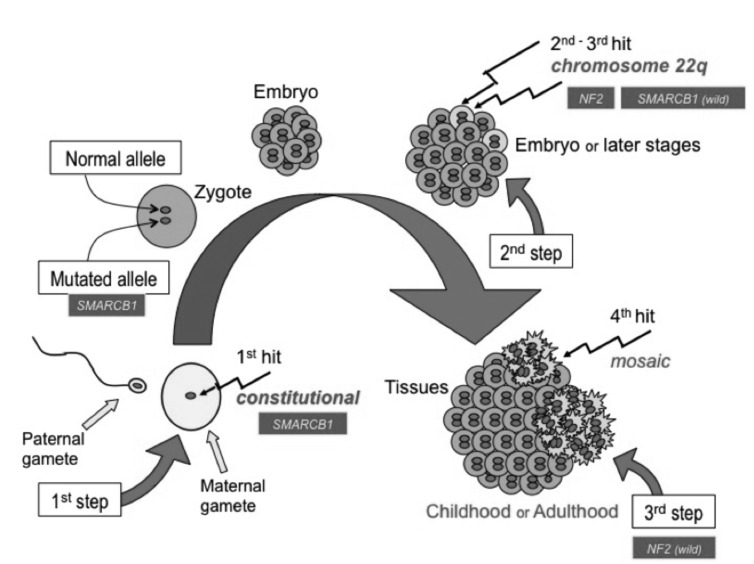
La neurofibromatosi tipo 2 [NF2] è una malattia genetica a trasmissione autosomica dominante [MIM # 101000]. Clinicamente è caratterizzata da: (1) schwannomi bilaterali del (VIII) nervo acustico/vestibolare; (2) cataratta giovanile o amartomi retinici; (3) schwannomi a carico dei nervi periferici e dei nervi cranici; (4) tumori multipli del sistema nervoso centrale (es., meningiomi, astrocitomi, ependimomi); (5) lesioni cutanee: (a) placche NF2 (schwannomi cutanei); (b) (poche) macchie caffellatte; (6) “malformazioni dello sviluppo corticale cerebrale”. La prevalenza della (forma sintomatica di) NF2 nella popolazione generale è di 1 su 100.000-200.000 individui con un’incidenza di 1 su 33.000 nati. La forma classica a esordio nel giovane adulto è conosciuta come forma di Gardner, (esordio intorno ai 20-30 anni d’età) con manifestazioni legate agli schwannomi bilaterali del nervo acustico/vestibolare (diminuzione/perdita progressiva dell’udito, tinnito, vertigini) e/o più raramente con manifestazioni da (altri) tumori del sistema nervoso centrale e/o periferico. In età pediatrica il fenotipo è diverso (forma di Wishart): per primi compaiono abitualmente i tumori del sistema nervoso centrale in assenza di schwannomi vestibolari; si possono avere macchie caffellatte e placche NF2 e solo dopo anni i tumori del nervo cranico VIII e di altri nervi cranici. Il quadro è più grave. Esiste anche una forma “congenita” ad esordio nei primi giorni/mesi di vita, con schwannomi vestibolari di piccole dimensioni (stabili nel tempo: anche per anni/decenni ma con improvvisa e rapida progressione) e numerose placche NF2; in questa forma le altre manifestazioni (es. meningiomi, altri tumori, altri schwannomi) sono spesso più gravi e progressive delle altre forme. Il gene responsabile della NF2 è localizzato sul cromosoma 22q12.1. Il prodotto genico della NF2 è conosciuto con il nome di schwannomina o merlina [dalla famiglia di proteine 4.1 del tipo moesina-ezrina-radixina/ERM alla quale appartiene il gene della NF2) e ha funzioni di regolazione della crescita e del rimodellamento cellulare (soppressione della crescita cellulare e della tumorigenesi)]. Alcune persone possono presentare tutte le (o parte delle) manifestazioni della NF2 in un emilato o in segmenti corporei circoscritti [NF2 a mosaico]. Altre persone presentano schwannomi (confermati istologicamente) dei nervi periferici (non intradermici) e/o delle radici gangliari in assenza di tumori del nervo vestibolare (o di altri nervi cranici: anche se in alcuni casi vi possono essere anche tumori unilaterali o bilaterali del nervo acustico/vestibolare e/o dei nervi cranici misti) o di altri segni diagnostici per la NF2 [Schwannomatosi, SWNTS]. L’esordio in questa forma è intorno ai 30 anni d’età (sono conosciuti casi in età pediatrica) con tumori in svariate sedi (abitualmente tronco e arti). Si conoscono due forme principali: (1) SWNTS1 [MIM # 162091] causata da alterazioni del gene SMARCB1 [regolatore della cromatina actina-dipendente associato alla matrice e correlato alle proteina SWI/SBF, sub-famiglia B, membro di tipo 1; MIM # 601607], sul cromosoma 22q11.23 (posizione centromerica rispetto al gene della NF2); (2) SWNTS2 [MIM # 615670] causata da alterazioni del gene LZTR1 [regolatore della trascrizione di tipo 1 legato alla Leucina; MIM # 600574], cromosoma 22q11.21 (posizione centromerica rispetto al gene SMARCB1) che codifica per una proteina, membro della super-famiglia BTB-kelch. Il meccanismo molecolare della Schwannomatosi comprende: (1) mutazione germinale del gene SMARCB1 o del gene LZTR1; (2) ampia delezione all’interno del cromosoma 22 (con perdita del gene NF2 e dell’allele intatto SMARCB1 o LZTR1); e (3) mutazione somatica dell’allele intatto del gene NF2 [meccanismo conosciuto come “four hits”: “Quadrupla alterazione” (su entrambi gli alleli dei due geni SWNTS/NF2), con tre passaggi consecutivi]. Negli ultimi anni, accanto alle tradizionali terapie chirurgiche e/o radioterapiche sono stati anche impiegati diversi farmaci “biologici” (es., Lapatinib e Bevacizumab) con effetti di riduzione/arresto della crescita dei tipici tumori NF2.
Keywords: Childhood NF2; Congenital NF2; Eearly onset NF2; Merlin; Mosaic NF2; Paediatric NF2; Schwannomatosis.
© Copyright by Società Italiana di Otorinolaringologia e Chirurgia Cervico-Facciale, Rome, Italy.
Figures
Similar articles
-
Diagnosis, Management, and New Therapeutic Options in Childhood Neurofibromatosis Type 2 and Related Forms.Semin Pediatr Neurol. 2015 Dec;22(4):240-58. doi: 10.1016/j.spen.2015.10.008. Epub 2015 Oct 28. Semin Pediatr Neurol. 2015. PMID: 26706012 Review.
-
A mosaic pattern of INI1/SMARCB1 protein expression distinguishes Schwannomatosis and NF2-associated peripheral schwannomas from solitary peripheral schwannomas and NF2-associated vestibular schwannomas.Childs Nerv Syst. 2017 Jun;33(6):933-940. doi: 10.1007/s00381-017-3340-2. Epub 2017 Apr 1. Childs Nerv Syst. 2017. PMID: 28365909
-
Neurofibromatosis type 2 (NF2): a clinical and molecular review.Orphanet J Rare Dis. 2009 Jun 19;4:16. doi: 10.1186/1750-1172-4-16. Orphanet J Rare Dis. 2009. PMID: 19545378 Free PMC article. Review.
-
[Neurofibromatosis type 2 (central neurofibromatosis or bilateral acoustic neuromas, vestibular schwannomas): from phenotype to gene].Lijec Vjesn. 2006 Sep-Oct;128(9-10):309-16. Lijec Vjesn. 2006. PMID: 17128670 Review. Croatian.
-
An update on the CNS manifestations of neurofibromatosis type 2.Acta Neuropathol. 2020 Apr;139(4):643-665. doi: 10.1007/s00401-019-02029-5. Epub 2019 Jun 4. Acta Neuropathol. 2020. PMID: 31161239 Free PMC article. Review.
Cited by
-
Social Independence of Patients with Neurofibromatosis Type 2 in Japan: Analysis of a National Registry of Patients Receiving Medical Expense Subsidies, 2004-2013.Neurol Med Chir (Tokyo). 2020 Sep 15;60(9):450-457. doi: 10.2176/nmc.oa.2020-0106. Epub 2020 Aug 15. Neurol Med Chir (Tokyo). 2020. PMID: 32801276 Free PMC article.
-
Splicing-Disrupting Mutations in Inherited Predisposition to Solid Pediatric Cancer.Cancers (Basel). 2022 Dec 2;14(23):5967. doi: 10.3390/cancers14235967. Cancers (Basel). 2022. PMID: 36497448 Free PMC article. Review.
-
Loss of social independence in patients with neurofibromatosis type 2: a follow-up study using a national registry in Japan.Environ Health Prev Med. 2023;28:46. doi: 10.1265/ehpm.22-00222. Environ Health Prev Med. 2023. PMID: 37599081 Free PMC article.
-
Rediagnosing one of Smith's patients (John McCann) with "neuromas tumours" (1849).Neurol Sci. 2017 Mar;38(3):493-499. doi: 10.1007/s10072-016-2797-1. Epub 2017 Jan 11. Neurol Sci. 2017. PMID: 28078568
-
Neurosurgical Treatment and Outcome of Pediatric Skull Base Lesions: A Case Series and Review of the Literature.Children (Basel). 2023 Jan 26;10(2):216. doi: 10.3390/children10020216. Children (Basel). 2023. PMID: 36832345 Free PMC article.
References
-
- Huson SM, Hughes RAC. A Pathogenetic and Clinical Overview. London: Churchill Livingstone; 1994. The Neurofibromatoses.
-
- Evans DG. Neurofibromatosis 2 Bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II. Genet Med. 2009;11:599–610. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
