

Identification of Small Molecule Compounds for Pharmacological Chaperone Therapy of Aspartylglucosaminuria
- PMID: 27876883
- PMCID: PMC5120323
- DOI: 10.1038/srep37583
Identification of Small Molecule Compounds for Pharmacological Chaperone Therapy of Aspartylglucosaminuria
Abstract
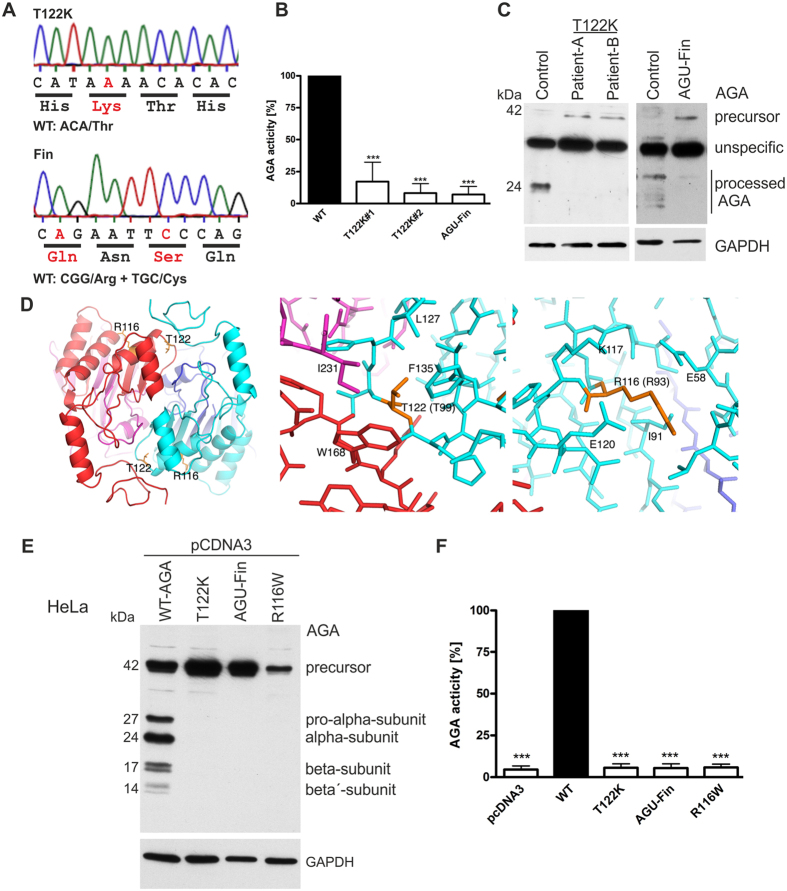
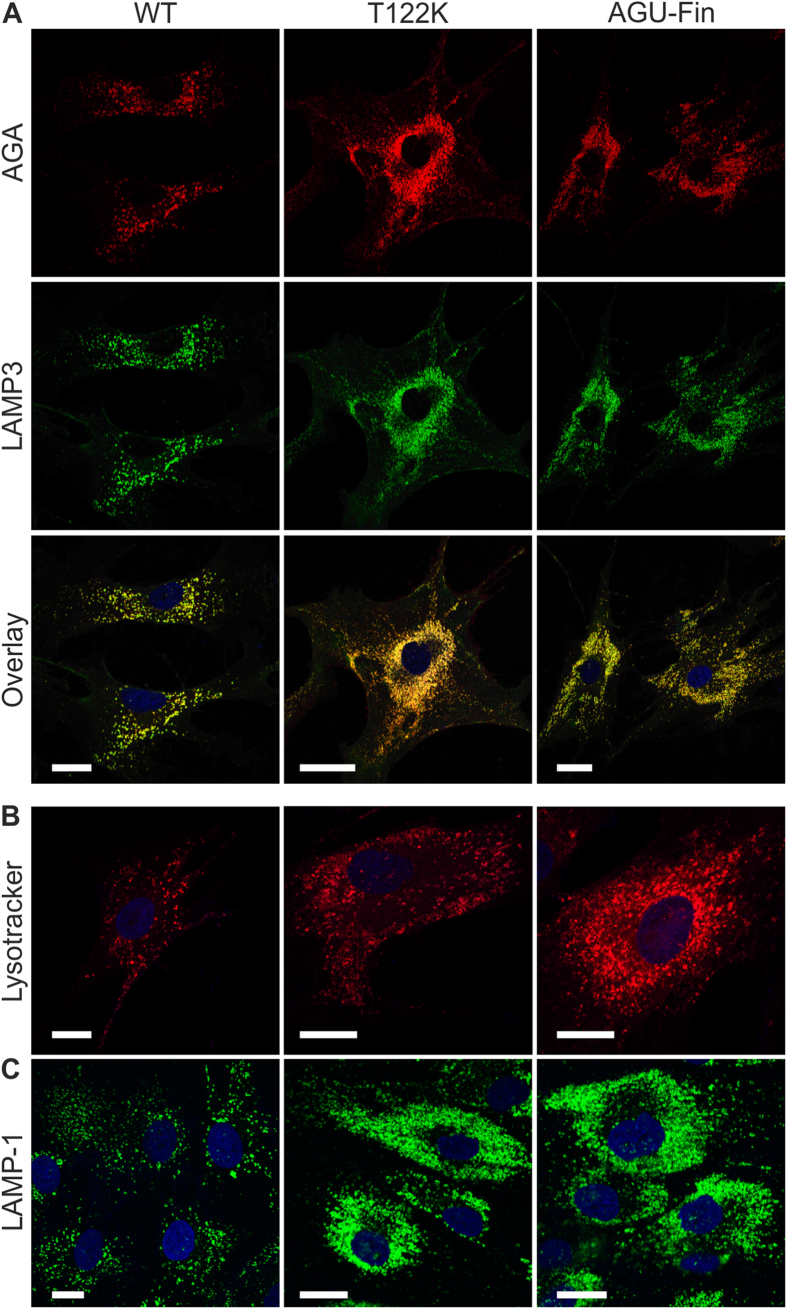
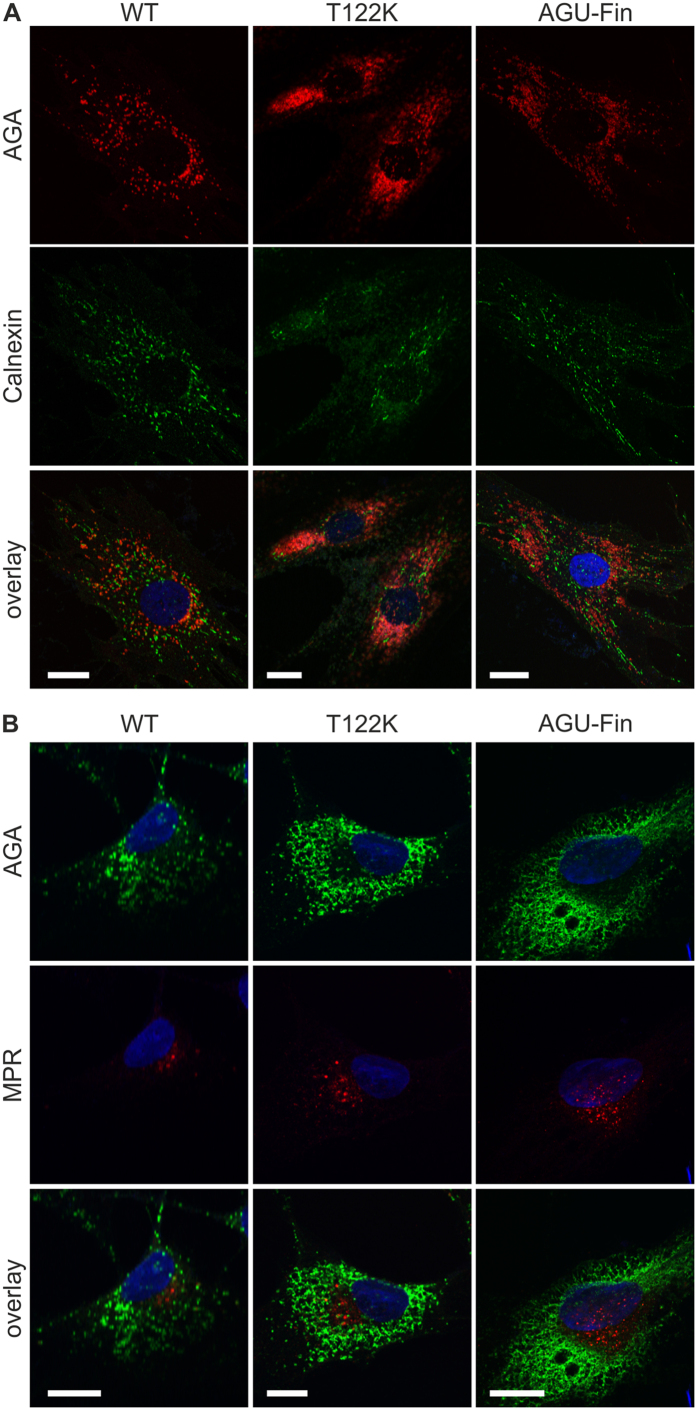
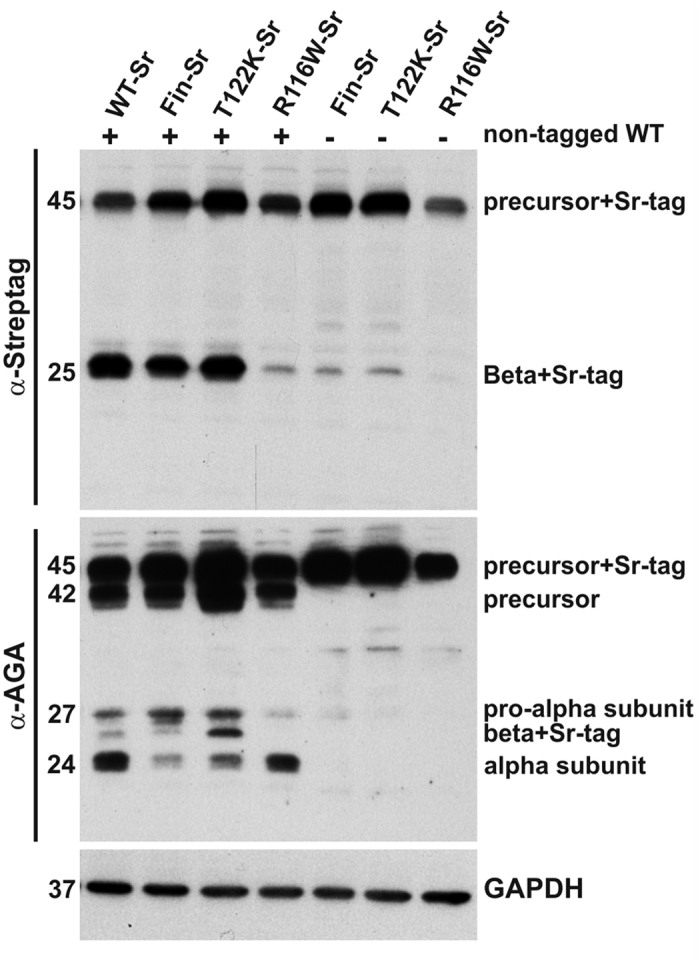
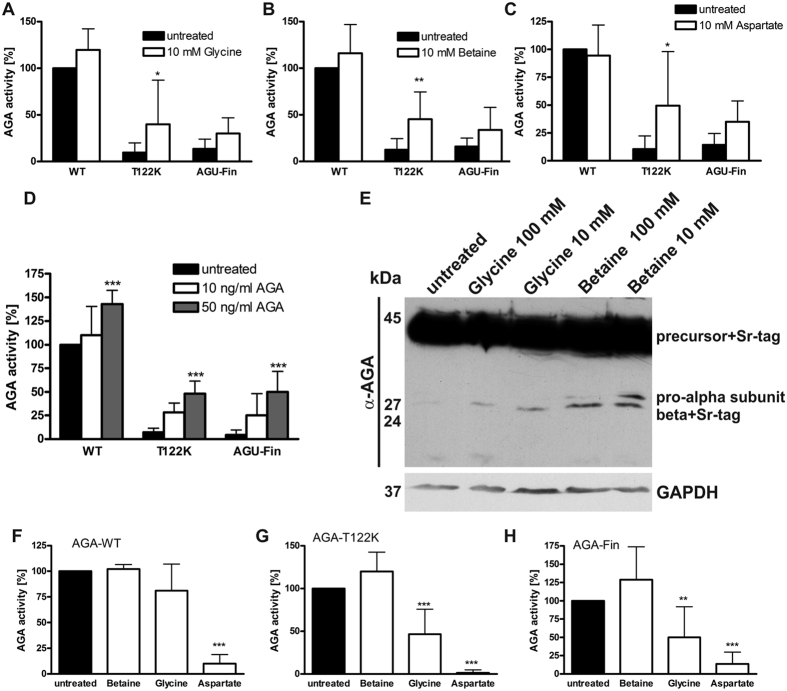
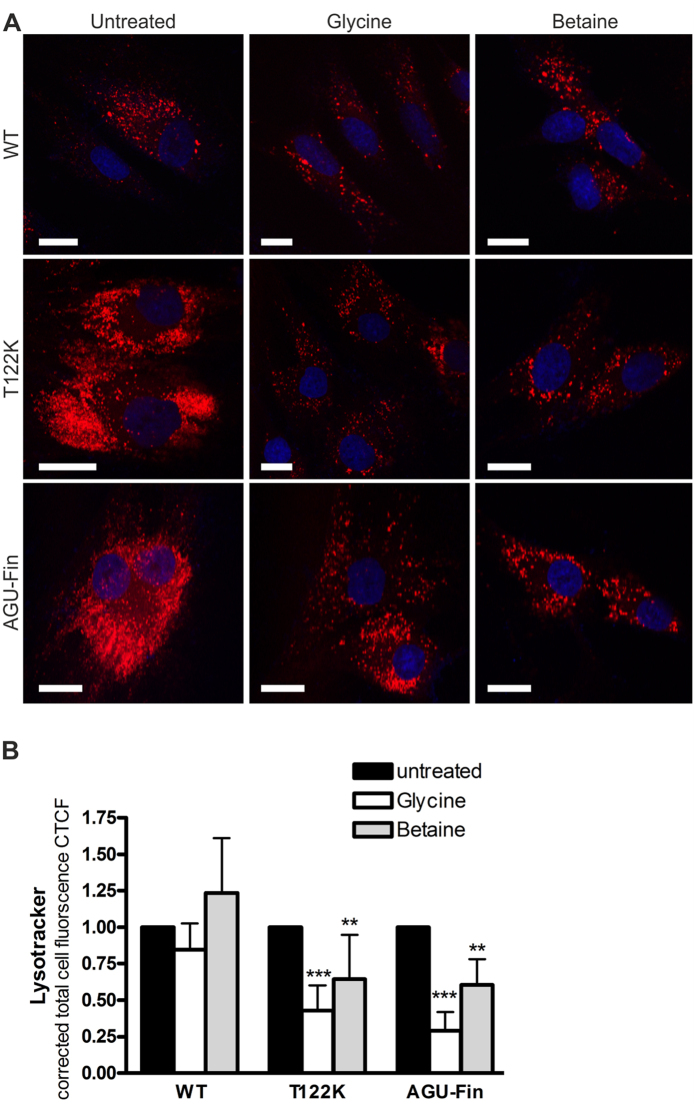
Aspartylglucosaminuria (AGU) is a lysosomal storage disorder that is caused by genetic deficiency of the enzyme aspartylglucosaminidase (AGA) which is involved in glycoprotein degradation. AGU is a progressive disorder that results in severe mental retardation in early adulthood. No curative therapy is currently available for AGU. We have here characterized the consequences of a novel AGU mutation that results in Thr122Lys exchange in AGA, and compared this mutant form to one carrying the worldwide most common AGU mutation, AGU-Fin. We show that T122K mutated AGA is expressed in normal amounts and localized in lysosomes, but exhibits low AGA activity due to impaired processing of the precursor molecule into subunits. Coexpression of T122K with wildtype AGA results in processing of the precursor into subunits, implicating that the mutation causes a local misfolding that prevents the precursor from becoming processed. Similar data were obtained for the AGU-Fin mutant polypeptide. We have here also identified small chemical compounds that function as chemical or pharmacological chaperones for the mutant AGA. Treatment of patient fibroblasts with these compounds results in increased AGA activity and processing, implicating that these substances may be suitable for chaperone mediated therapy for AGU.
Figures
Similar articles
-
Amlexanox provides a potential therapy for nonsense mutations in the lysosomal storage disorder Aspartylglucosaminuria.Biochim Biophys Acta Mol Basis Dis. 2018 Mar;1864(3):668-675. doi: 10.1016/j.bbadis.2017.12.014. Epub 2017 Dec 13. Biochim Biophys Acta Mol Basis Dis. 2018. PMID: 29247835
-
Molecular pathogenesis of a disease: structural consequences of aspartylglucosaminuria mutations.Hum Mol Genet. 2001 Apr 15;10(9):983-95. doi: 10.1093/hmg/10.9.983. Hum Mol Genet. 2001. PMID: 11309371
-
A novel aspartylglucosaminuria mutation affects translocation of aspartylglucosaminidase.Hum Mutat. 2004 Oct;24(4):350-1. doi: 10.1002/humu.9276. Hum Mutat. 2004. PMID: 15365992
-
Aspartylglycosaminuria: a review.Orphanet J Rare Dis. 2016 Dec 1;11(1):162. doi: 10.1186/s13023-016-0544-6. Orphanet J Rare Dis. 2016. PMID: 27906067 Free PMC article. Review.
-
Dissection of the molecular pathology of aspartylglucosaminuria provides the basis for DNA diagnostics and future therapeutic interventions.Scand J Clin Lab Invest Suppl. 1993;213:19-27. doi: 10.3109/00365519309090670. Scand J Clin Lab Invest Suppl. 1993. PMID: 8322015 Review.
Cited by
-
The Finnish genetic heritage in 2022 - from diagnosis to translational research.Dis Model Mech. 2022 Oct 1;15(10):dmm049490. doi: 10.1242/dmm.049490. Epub 2022 Oct 26. Dis Model Mech. 2022. PMID: 36285626 Free PMC article. Review.
-
Lysosomal diseases: Overview on current diagnosis and treatment.Genet Mol Biol. 2019;42(1 suppl 1):165-177. doi: 10.1590/1678-4685-GMB-2018-0159. Epub 2019 Apr 25. Genet Mol Biol. 2019. PMID: 31067291 Free PMC article.
-
E-Learning for Rare Diseases: An Example Using Fabry Disease.Int J Mol Sci. 2017 Sep 24;18(10):2049. doi: 10.3390/ijms18102049. Int J Mol Sci. 2017. PMID: 28946642 Free PMC article.
-
Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses.Int J Mol Sci. 2019 Dec 29;21(1):232. doi: 10.3390/ijms21010232. Int J Mol Sci. 2019. PMID: 31905715 Free PMC article. Review.
-
Functional Analysis of the Ser149/Thr149 Variants of Human Aspartylglucosaminidase and Optimization of the Coding Sequence for Protein Production.Int J Mol Sci. 2017 Mar 26;18(4):706. doi: 10.3390/ijms18040706. Int J Mol Sci. 2017. PMID: 28346360 Free PMC article.
References
-
- Opladen T. et al.. Aspartylglucosaminuria: unusual neonatal presentation in Qatari twins with a novel aspartylglucosaminidase gene mutation and 3 new cases in a Turkish family. J Child Neurol 29, 36–42 (2014). - PubMed
-
- Saarela J. et al.. Molecular pathogenesis of a disease: structural consequences of aspartylglucosaminuria mutations. Hum Mol Genet 10, 983–995 (2001). - PubMed
-
- Tikkanen R., Enomaa N., Riikonen A., Ikonen E. & Peltonen L. Intracellular sorting of aspartylglucosaminidase: the role of N-linked oligosaccharides and evidence of Man-6-P-independent lysosomal targeting. DNA Cell Biol 14, 305–312 (1995). - PubMed
-
- Oinonen C., Tikkanen R., Rouvinen J. & Peltonen L. Three-dimensional structure of human lysosomal aspartylglucosaminidase. Nat Struct Biol 2, 1102–1108 (1995). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
