

The role of genetics in pulmonary arterial hypertension
- PMID: 27770446
- PMCID: PMC5167647
- DOI: 10.1002/path.4833
The role of genetics in pulmonary arterial hypertension
Abstract
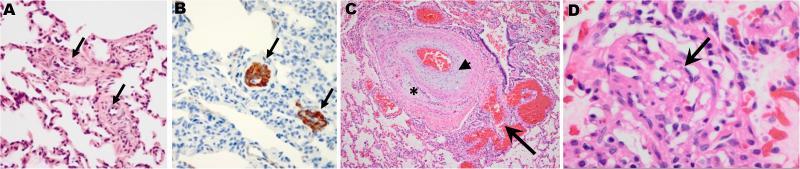
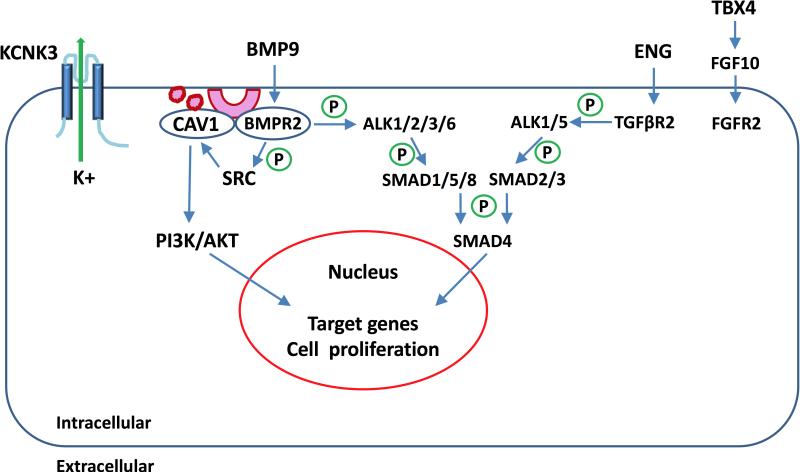
Group 1 pulmonary hypertension or pulmonary arterial hypertension (PAH) is a rare disease characterized by proliferation and occlusion of small pulmonary arterioles, leading to progressive elevation of pulmonary artery pressure and pulmonary vascular resistance, and right ventricular failure. Historically, it has been associated with a high mortality rate, although, over the last decade, treatment has improved survival. PAH includes idiopathic PAH (IPAH), heritable PAH (HPAH), and PAH associated with certain medical conditions. The aetiology of PAH is heterogeneous, and genetics play an important role in some cases. Mutations in BMPR2, encoding bone morphogenetic protein receptor 2, a member of the transforming growth factor-β superfamily of receptors, have been identified in 70% of cases of HPAH, and in 10-40% of cases of IPAH. Other genetic causes of PAH include mutations in the genes encoding activin receptor-like type 1, endoglin, SMAD9, caveolin 1, and potassium two-pore-domain channel subfamily K member 3. Mutations in the gene encoding T-box 4 have been identified in 10-30% of paediatric PAH patients, but rarely in adults with PAH. PAH in children is much more heterogeneous than in adults, and can be associated with several genetic syndromes, congenital heart disease, pulmonary disease, and vascular disease. In addition to rare mutations as a monogenic cause of HPAH, common variants in the gene encoding cerebellin 2 increase the risk of PAH by approximately two-fold. A PAH panel of genes is available for clinical testing, and should be considered for use in clinical management, especially for patients with a family history of PAH. Copyright © 2016 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Keywords: BMPR2; CAV1; KCNK3; TBX4; TGF-β; genetics.
Copyright © 2016 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Figures
Similar articles
-
The genetic basis of pulmonary arterial hypertension.Hum Genet. 2014 May;133(5):471-9. doi: 10.1007/s00439-014-1419-3. Epub 2014 Jan 21. Hum Genet. 2014. PMID: 24442418 Review.
-
EIF2AK4 Mutations in Patients Diagnosed With Pulmonary Arterial Hypertension.Chest. 2017 Apr;151(4):821-828. doi: 10.1016/j.chest.2016.11.014. Epub 2016 Nov 22. Chest. 2017. PMID: 27884767
-
Genetics of pulmonary arterial hypertension.Clin Chest Med. 2013 Dec;34(4):651-63. doi: 10.1016/j.ccm.2013.08.003. Clin Chest Med. 2013. PMID: 24267296 Review.
-
Pulmonary artery hypertension in childhood: The transforming growth factor-β superfamily-related genes.Pediatr Neonatol. 2018 Apr;59(2):112-119. doi: 10.1016/j.pedneo.2016.12.008. Epub 2017 Aug 12. Pediatr Neonatol. 2018. PMID: 28967497 Review.
-
Bmpr2 Mutant Rats Develop Pulmonary and Cardiac Characteristics of Pulmonary Arterial Hypertension.Circulation. 2019 Feb 12;139(7):932-948. doi: 10.1161/CIRCULATIONAHA.118.033744. Circulation. 2019. PMID: 30586714
Cited by
-
Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension.Genome Med. 2019 Nov 14;11(1):69. doi: 10.1186/s13073-019-0685-z. Genome Med. 2019. PMID: 31727138 Free PMC article.
-
Lung pathology for the clinician: a comprehensive approach.Eur Respir Rev. 2017 Jun 28;26(144):170041. doi: 10.1183/16000617.0041-2017. Print 2017 Jun 30. Eur Respir Rev. 2017. PMID: 28659505 Free PMC article.
-
The effect of the mechanodynamic lung environment on fibroblast phenotype via the Flexcell.BMC Pulm Med. 2024 Jul 27;24(1):362. doi: 10.1186/s12890-024-03167-7. BMC Pulm Med. 2024. PMID: 39068387 Free PMC article. Review.
-
A Combined Targeted and Whole Exome Sequencing Approach Identified Novel Candidate Genes Involved in Heritable Pulmonary Arterial Hypertension.Sci Rep. 2019 Jan 24;9(1):753. doi: 10.1038/s41598-018-37277-0. Sci Rep. 2019. PMID: 30679663 Free PMC article.
-
TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family.Int J Mol Sci. 2018 Aug 31;19(9):2585. doi: 10.3390/ijms19092585. Int J Mol Sci. 2018. PMID: 30200294 Free PMC article. Review.
References
-
- Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–163. - PubMed
-
- Lane KB, Machado RD, Pauciulo MW, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26:81–84. - PubMed
-
- Aldred MA, Vijayakrishnan J, James V, et al. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum Mutat. 2006;27:212–213. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
