

Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing
- PMID: 27479907
- PMCID: PMC5988037
- DOI: 10.1038/ng.3627
Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing
Abstract
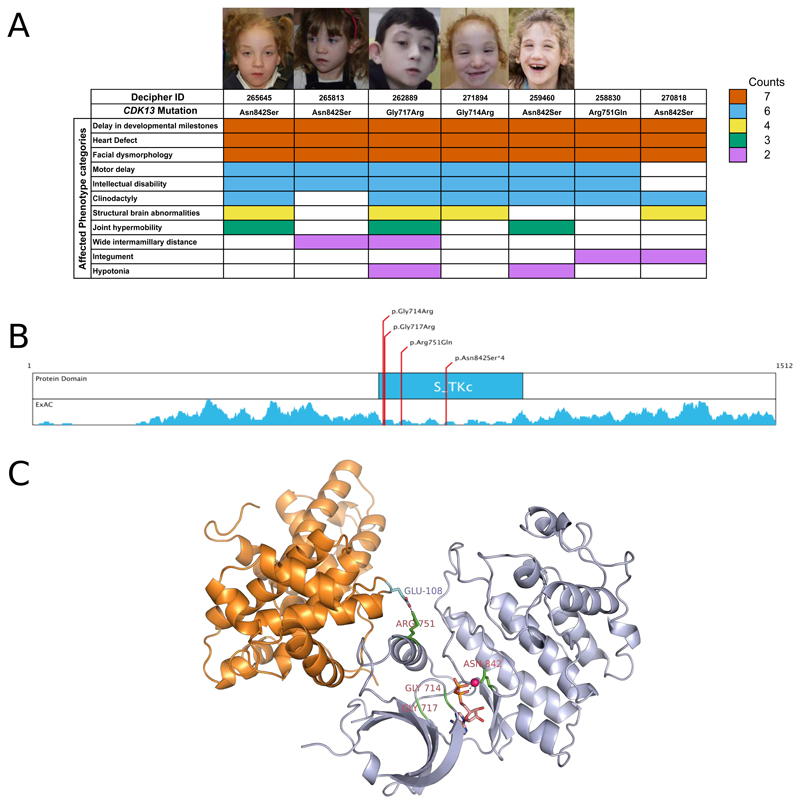
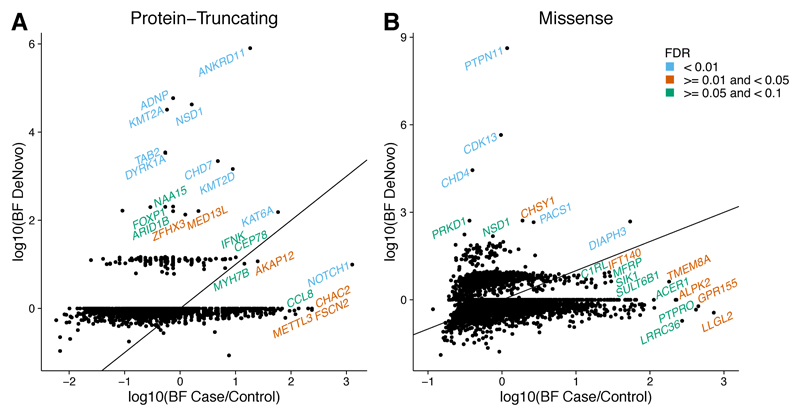
Congenital heart defects (CHDs) have a neonatal incidence of 0.8-1% (refs. 1,2). Despite abundant examples of monogenic CHD in humans and mice, CHD has a low absolute sibling recurrence risk (∼2.7%), suggesting a considerable role for de novo mutations (DNMs) and/or incomplete penetrance. De novo protein-truncating variants (PTVs) have been shown to be enriched among the 10% of 'syndromic' patients with extra-cardiac manifestations. We exome sequenced 1,891 probands, including both syndromic CHD (S-CHD, n = 610) and nonsyndromic CHD (NS-CHD, n = 1,281). In S-CHD, we confirmed a significant enrichment of de novo PTVs but not inherited PTVs in known CHD-associated genes, consistent with recent findings. Conversely, in NS-CHD we observed significant enrichment of PTVs inherited from unaffected parents in CHD-associated genes. We identified three genome-wide significant S-CHD disorders caused by DNMs in CHD4, CDK13 and PRKD1. Our study finds evidence for distinct genetic architectures underlying the low sibling recurrence risk in S-CHD and NS-CHD.
Conflict of interest statement
M.E.H. is a co-founder of, and holds shares in, Congenica Ltd, a genetics diagnostic company.
Figures


Similar articles
-
De novo damaging variants associated with congenital heart diseases contribute to the connectome.Sci Rep. 2020 Apr 27;10(1):7046. doi: 10.1038/s41598-020-63928-2. Sci Rep. 2020. PMID: 32341405 Free PMC article.
-
Effect of deletion of the protein kinase PRKD1 on development of the mouse embryonic heart.J Anat. 2024 Jul;245(1):70-83. doi: 10.1111/joa.14033. Epub 2024 Feb 28. J Anat. 2024. PMID: 38419169 Free PMC article.
-
De novo variants in exomes of congenital heart disease patients identify risk genes and pathways.Genome Med. 2020 Jan 15;12(1):9. doi: 10.1186/s13073-019-0709-8. Genome Med. 2020. PMID: 31941532 Free PMC article.
-
What Is New in Genetics of Congenital Heart Defects?Front Pediatr. 2016 Dec 1;4:120. doi: 10.3389/fped.2016.00120. eCollection 2016. Front Pediatr. 2016. PMID: 27990414 Free PMC article. Review.
-
Complex genetics and the etiology of human congenital heart disease.Cold Spring Harb Perspect Med. 2014 Jul 1;4(7):a013953. doi: 10.1101/cshperspect.a013953. Cold Spring Harb Perspect Med. 2014. PMID: 24985128 Free PMC article. Review.
Cited by
-
Functional testing of a human PBX3 variant in zebrafish reveals a potential modifier role in congenital heart defects.Dis Model Mech. 2018 Oct 18;11(10):dmm035972. doi: 10.1242/dmm.035972. Dis Model Mech. 2018. PMID: 30355621 Free PMC article.
-
The genetics of isolated congenital heart disease.Am J Med Genet C Semin Med Genet. 2020 Mar;184(1):97-106. doi: 10.1002/ajmg.c.31763. Epub 2019 Dec 26. Am J Med Genet C Semin Med Genet. 2020. PMID: 31876989 Free PMC article. Review.
-
Micro-RNA signatures in monozygotic twins discordant for congenital heart defects.PLoS One. 2019 Dec 5;14(12):e0226164. doi: 10.1371/journal.pone.0226164. eCollection 2019. PLoS One. 2019. PMID: 31805172 Free PMC article.
-
Genetic architecture of laterality defects revealed by whole exome sequencing.Eur J Hum Genet. 2019 Apr;27(4):563-573. doi: 10.1038/s41431-018-0307-z. Epub 2019 Jan 8. Eur J Hum Genet. 2019. PMID: 30622330 Free PMC article.
-
Exploring the Genetic Architecture of Spontaneous Coronary Artery Dissection Using Whole-Genome Sequencing.Circ Genom Precis Med. 2022 Aug;15(4):e003527. doi: 10.1161/CIRCGEN.121.003527. Epub 2022 May 18. Circ Genom Precis Med. 2022. PMID: 35583931 Free PMC article.
References
-
- Hoffman JIE, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–900. - PubMed
-
- Øyen N, et al. Recurrence of congenital heart defects in families. Circulation. 2009;120:295–301. - PubMed
-
- Gill HK, Splitt M, Sharland GK, Simpson JM. Patterns of recurrence of congenital heart disease: an analysis of 6,640 consecutive pregnancies evaluated by detailed fetal echocardiography. J Am Coll Cardiol. 2003;42:923–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RP-PG-0310-1004/DH_/Department of Health/United Kingdom
- RG/13/13/30194/BHF_/British Heart Foundation/United Kingdom
- RG/10/17/28553/BHF_/British Heart Foundation/United Kingdom
- RG/15/12/31616/BHF_/British Heart Foundation/United Kingdom
- FS/14/51/30879/BHF_/British Heart Foundation/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- RP-PG-0310-1002/DH_/Department of Health/United Kingdom
- RG/09/012/28096/BHF_/British Heart Foundation/United Kingdom
- PG/07/045/22690/BHF_/British Heart Foundation/United Kingdom
- MC_PC_U127561093/MRC_/Medical Research Council/United Kingdom
- RG/13/10/30376/BHF_/British Heart Foundation/United Kingdom
- MR/L003120/1/MRC_/Medical Research Council/United Kingdom
- 091986/WT_/Wellcome Trust/United Kingdom
- RG/07/010/23676/BHF_/British Heart Foundation/United Kingdom
- MC_PC_15018/MRC_/Medical Research Council/United Kingdom
- 098051/WT_/Wellcome Trust/United Kingdom
- CIHR/Canada
- SP/09/002/BHF_/British Heart Foundation/United Kingdom
- WT098051/WT_/Wellcome Trust/United Kingdom
- G0800270/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
