

Clinical and neuroradiological features of spinocerebellar ataxia 38 (SCA38)
- PMID: 27143115
- PMCID: PMC4925464
- DOI: 10.1016/j.parkreldis.2016.04.030
Clinical and neuroradiological features of spinocerebellar ataxia 38 (SCA38)
Abstract
Introduction: SCA38 (MIM 611805) caused by mutations within the ELOVL5 gene, which encodes an enzyme involved in the synthesis of long-chain fatty acids with a high and specific expression in Purkinje cells, has recently been identified.
Objective: The present study was aimed at describing the clinical and neuroimaging features, and the natural history of SCA38.
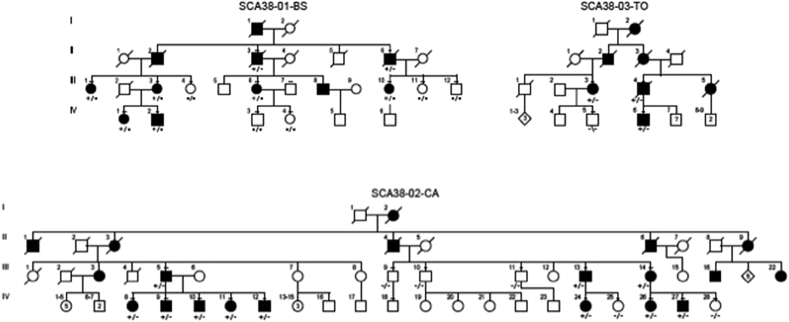
Methods: We extended our clinical and brain neuroimaging data on SCA38 including 21 cases from three Italian families. All had the ELOVL5 c.689G > T (p.Gly230Val) missense mutation.
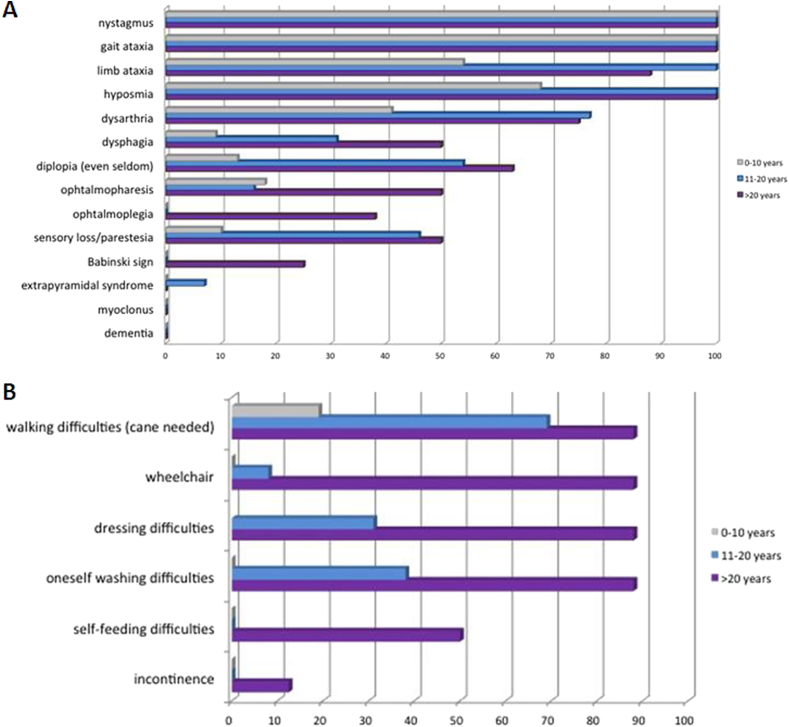
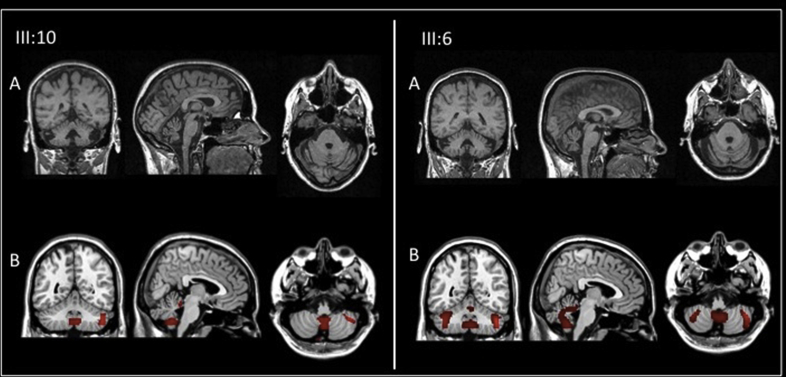
Results: Age at disease onset was in the fourth decade of life. The presenting features were nystagmus (100% of cases) and slowly progressive gait ataxia (95%). Frequent signs and symptoms included pes cavus (82%) and hyposmia (76%); rarer symptoms were hearing loss (33%) and anxiety disorder (33%). The disease progressed with cerebellar symptoms such as limb ataxia, dysarthria, dysphagia, and ophtalmoparesis followed in the later stages by ophtalmoplegia. Peripheral nervous system involvement was present in the last phase of disease with sensory loss. Dementia or extrapyramidal signs were not detected. Significant loss of abilities of daily living was reported only after 20 years of the disease. Brain imaging documented cerebellar atrophy with sparing of cerebral cortex and no white matter disease.
Conclusions: SCA38 is a rare form of inherited ataxia with characteristic clinical features, including pes cavus and hyposmia, that may guide genetic screening and prompt diagnosis in light of possible future therapeutic interventions.
Keywords: Ataxia; ELOVL5; Gene; Mutation; SCA38.
Copyright © 2016 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures
Similar articles
-
Effects of the administration of Elovl5-dependent fatty acids on a spino-cerebellar ataxia 38 mouse model.Behav Brain Funct. 2022 Aug 6;18(1):8. doi: 10.1186/s12993-022-00194-4. Behav Brain Funct. 2022. PMID: 35933444 Free PMC article.
-
ELOVL5 mutations cause spinocerebellar ataxia 38.Am J Hum Genet. 2014 Aug 7;95(2):209-17. doi: 10.1016/j.ajhg.2014.07.001. Epub 2014 Jul 24. Am J Hum Genet. 2014. PMID: 25065913 Free PMC article.
-
Spinocerebellar ataxia 38: structure-function analysis shows ELOVL5 G230V is proteotoxic, conformationally altered and a mutational hotspot.Hum Genet. 2023 Aug;142(8):1055-1076. doi: 10.1007/s00439-023-02572-y. Epub 2023 May 18. Hum Genet. 2023. PMID: 37199746 Free PMC article.
-
Spinocerebellar ataxia type 15.Cerebellum. 2005;4(1):47-50. doi: 10.1080/14734220410019029. Cerebellum. 2005. PMID: 15895559 Review.
-
Spinocerebellar ataxia type 31: A clinical and radiological literature review.J Neurol Sci. 2023 Jan 15;444:120527. doi: 10.1016/j.jns.2022.120527. Epub 2022 Dec 16. J Neurol Sci. 2023. PMID: 36563608 Review.
Cited by
-
A Systematic Review of the Spectrum and Prevalence of Non-Motor Symptoms in Adults with Hereditary Cerebellar Ataxias.Mov Disord Clin Pract. 2022 Sep 6;9(8):1027-1039. doi: 10.1002/mdc3.13532. eCollection 2022 Nov. Mov Disord Clin Pract. 2022. PMID: 36339305 Free PMC article. Review.
-
Neuroradiological Findings in the Spinocerebellar Ataxias.Tremor Other Hyperkinet Mov (N Y). 2019 Sep 26;9. doi: 10.7916/tohm.v0.682. eCollection 2019. Tremor Other Hyperkinet Mov (N Y). 2019. PMID: 31632837 Free PMC article. Review.
-
Effects of the administration of Elovl5-dependent fatty acids on a spino-cerebellar ataxia 38 mouse model.Behav Brain Funct. 2022 Aug 6;18(1):8. doi: 10.1186/s12993-022-00194-4. Behav Brain Funct. 2022. PMID: 35933444 Free PMC article.
-
Lipid Dyshomeostasis and Inherited Cerebellar Ataxia.Mol Neurobiol. 2022 Jun;59(6):3800-3828. doi: 10.1007/s12035-022-02826-2. Epub 2022 Apr 14. Mol Neurobiol. 2022. PMID: 35420383 Free PMC article. Review.
-
A Review of Ocular Movement Abnormalities in Hereditary Cerebellar Ataxias.Cerebellum. 2024 Apr;23(2):702-721. doi: 10.1007/s12311-023-01554-0. Epub 2023 Mar 31. Cerebellum. 2024. PMID: 37000369 Review.
References
-
- Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–894. - PubMed
-
- Schöls L., Bauer P., Schmidt T., Schulte T., Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. - PubMed
-
- Globas C., du Montcel S.T., Baliko L., Boesch S., Depondt C., DiDonato S., Durr A., Filla A., Klockgether T., Mariotti C., Melegh B., Rakowicz M., Ribai P., Rola R., Schmitz-Hubsch T., Szymanski S., Timmann D., Van de Warrenburg B.P., Bauer P., Schols L. Early symptoms in spinocerebellar ataxia type 1, 2, 3, and 6. Mov. Disord. 2008;23:2232–2238. - PubMed
-
- Yamada M., Sato T., Tsuji S., Takahashi H. CAG repeat disorder models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:71–86. - PubMed
-
- Rossi M., Perez-Lloret S., Doldan L., Cerquetti D., Balej J., Millar Vernetti P., Hawkes H., Cammarota A., Merello M. Autosomal dominant cerebellar ataxias: a systematic review of clinical features. Eur. J. Neurol. 2014;21:607–615. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
