

Perspective on the Genetics and Diagnosis of Congenital Hyperinsulinism Disorders
- PMID: 26908106
- PMCID: PMC4803157
- DOI: 10.1210/jc.2015-3651
Perspective on the Genetics and Diagnosis of Congenital Hyperinsulinism Disorders
Abstract
Context: Congenital hyperinsulinism (HI) is the most common cause of hypoglycemia in children. The risk of permanent brain injury in infants with HI continues to be as high as 25-50% due to delays in diagnosis and inadequate treatment. Congenital HI has been described since the birth of the JCEM under various terms, including "idiopathic hypoglycemia of infancy," "leucine-sensitive hypoglycemia," or "nesidioblastosis."
Evidence acquisition: In the past 20 years, it has become apparent that HI is caused by genetic defects in the pathways that regulate pancreatic β-cell insulin secretion.
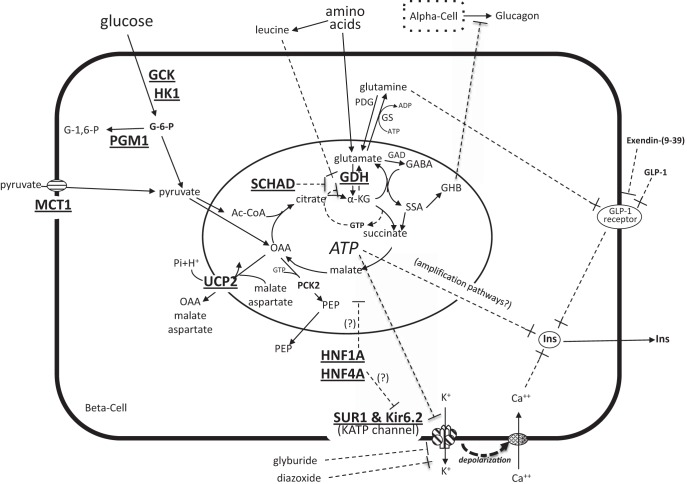
Evidence synthesis: There are now 11 genes associated with monogenic forms of HI (ABCC8, KCNJ11, GLUD1, GCK, HADH1, UCP2, MCT1, HNF4A, HNF1A, HK1, PGM1), as well as several syndromic genetic forms of HI (eg, Beckwith-Wiedemann, Kabuki, and Turner syndromes). HI is also the cause of hypoglycemia in transitional neonatal hypoglycemia and in persistent hypoglycemia in various groups of high-risk neonates (such as birth asphyxia, small for gestational age birthweight, infant of diabetic mother). Management of HI is one of the most difficult problems faced by pediatric endocrinologists and frequently requires difficult choices, such as near-total pancreatectomy and/or highly intensive care with continuous tube feedings. For 50 years, diazoxide, a KATP channel agonist, has been the primary drug for infants with HI; however, it is ineffective in most cases with mutations of ABCC8 or KCNJ11, which constitute the majority of infants with monogenic HI.
Conclusions: Genetic mutation testing has become standard of care for infants with HI and has proven to be useful not only in projecting prognosis and family counseling, but also in diagnosing infants with surgically curable focal HI lesions. (18)F-fluoro-L-dihydroxyphenylalanine ((18)F-DOPA) PET scans have been found to be highly accurate for localizing such focal lesions preoperatively. New drugs under investigation provide hope for improving the outcomes of children with HI.
Figures
Similar articles
-
Genotype and phenotype correlations in 417 children with congenital hyperinsulinism.J Clin Endocrinol Metab. 2013 Feb;98(2):E355-63. doi: 10.1210/jc.2012-2169. Epub 2012 Dec 28. J Clin Endocrinol Metab. 2013. PMID: 23275527 Free PMC article.
-
Hyperinsulinism in the Neonate.Clin Perinatol. 2018 Mar;45(1):61-74. doi: 10.1016/j.clp.2017.10.007. Epub 2017 Dec 6. Clin Perinatol. 2018. PMID: 29406007 Review.
-
Congenital hyperinsulinism.Early Hum Dev. 2010 May;86(5):287-94. doi: 10.1016/j.earlhumdev.2010.05.003. Epub 2010 Jun 13. Early Hum Dev. 2010. PMID: 20550977 Review.
-
Congenital Hyperinsulinemic Hypoglycemia and Hyperammonemia due to Pathogenic Variants in GLUD1.Indian J Pediatr. 2019 Nov;86(11):1051-1053. doi: 10.1007/s12098-019-02980-x. Epub 2019 May 22. Indian J Pediatr. 2019. PMID: 31119523
-
Hyperinsulinism in infancy and childhood: when an insulin level is not always enough.Clin Chem. 2008 Feb;54(2):256-63. doi: 10.1373/clinchem.2007.098988. Epub 2007 Dec 21. Clin Chem. 2008. PMID: 18156285 Review.
Cited by
-
Adult focal β-cell nesidioblastosis: A case report.World J Clin Cases. 2023 Jan 6;11(1):150-156. doi: 10.12998/wjcc.v11.i1.150. World J Clin Cases. 2023. PMID: 36687197 Free PMC article.
-
Hypoglycaemia Metabolic Gene Panel Testing.Front Endocrinol (Lausanne). 2022 Mar 29;13:826167. doi: 10.3389/fendo.2022.826167. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35422763 Free PMC article. Review.
-
Investigating Genetic Mutations in a Large Cohort of Iranian Patients with Congenital Hyperinsulinism.J Clin Res Pediatr Endocrinol. 2022 Mar 3;14(1):87-95. doi: 10.4274/jcrpe.galenos.2021.2021.0071. Epub 2021 Dec 20. J Clin Res Pediatr Endocrinol. 2022. PMID: 34927408 Free PMC article.
-
The Genetic and Molecular Mechanisms of Congenital Hyperinsulinism.Front Endocrinol (Lausanne). 2019 Feb 26;10:111. doi: 10.3389/fendo.2019.00111. eCollection 2019. Front Endocrinol (Lausanne). 2019. PMID: 30873120 Free PMC article. Review.
-
Using referral rates for genetic testing to determine the incidence of a rare disease: The minimal incidence of congenital hyperinsulinism in the UK is 1 in 28,389.PLoS One. 2020 Feb 6;15(2):e0228417. doi: 10.1371/journal.pone.0228417. eCollection 2020. PLoS One. 2020. PMID: 32027664 Free PMC article.
References
-
- Hartmann AF, Jaudon JC. Hypoglycemia. J Pediatr. 1937;11:1–36.
-
- McQuarrie I. Idiopathic spontaneously occurring hypoglycemia in infants; clinical significance of problem and treatment. AMA Am J Dis Child. 1954;87:399–428. - PubMed
-
- Stanley CA, Baker L. Hyperinsulinism in infants and children: diagnosis and therapy. Adv Pediatr. 1976;23:315–355. - PubMed
-
- Pagliara AS, Karl IE, Haymond M, Kipnis DM. Hypoglycemia in infancy and childhood. II. J Pediatr. 1973;82:558–577. - PubMed
-
- Steinkrauss L, Lipman TH, Hendell CD, Gerdes M, Thornton PS, Stanley CA. Effects of hypoglycemia on developmental outcome in children with congenital hyperinsulinism. J Pediatr Nurs. 2005;20:109–118. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
