

Novel SLC19A3 Promoter Deletion and Allelic Silencing in Biotin-Thiamine-Responsive Basal Ganglia Encephalopathy
- PMID: 26863430
- PMCID: PMC4749299
- DOI: 10.1371/journal.pone.0149055
Novel SLC19A3 Promoter Deletion and Allelic Silencing in Biotin-Thiamine-Responsive Basal Ganglia Encephalopathy
Abstract
Background: Biotin-thiamine responsive basal ganglia disease is a severe, but potentially treatable disorder caused by mutations in the SLC19A3 gene. Although the disease is inherited in an autosomal recessive manner, patients with typical phenotypes carrying single heterozygous mutations have been reported. This makes the diagnosis uncertain and may delay treatment.
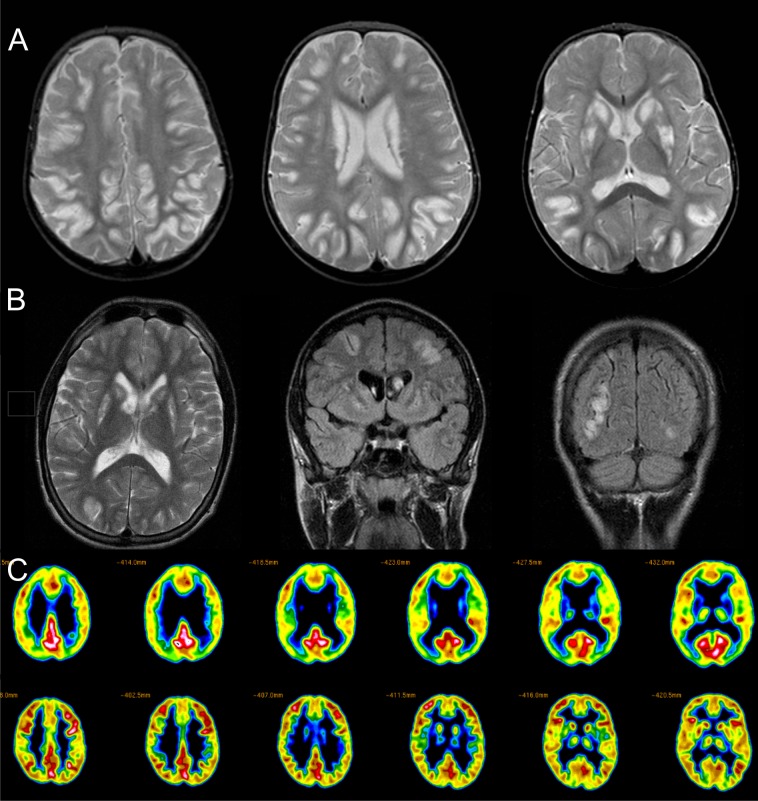
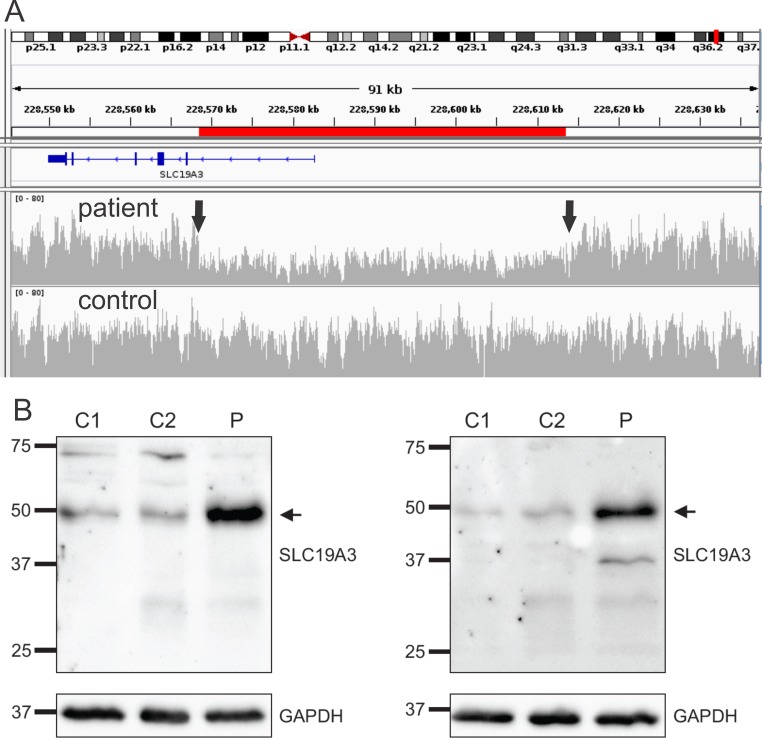
Methods and results: In two siblings with early-onset encephalopathy dystonia and epilepsy, whole-exome sequencing revealed a novel single heterozygous SLC19A3 mutation (c.337T>C). Although Sanger-sequencing and copy-number analysis revealed no other aberrations, RNA-sequencing in brain tissue suggested the second allele was silenced. Whole-genome sequencing resolved the genetic defect by revealing a novel 45,049 bp deletion in the 5'-UTR region of the gene abolishing the promoter. High dose thiamine and biotin therapy was started in the surviving sibling who remains stable. In another patient two novel compound heterozygous SLC19A3 mutations were found. He improved substantially on thiamine and biotin therapy.
Conclusions: We show that large genomic deletions occur in the regulatory region of SLC19A3 and should be considered in genetic testing. Moreover, our study highlights the power of whole-genome sequencing as a diagnostic tool for rare genetic disorders across a wide spectrum of mutations including non-coding large genomic rearrangements.
Conflict of interest statement
Figures
Similar articles
-
Compound heterozygous SLC19A3 mutations further refine the critical promoter region for biotin-thiamine-responsive basal ganglia disease.Cold Spring Harb Mol Case Stud. 2017 Nov 21;3(6):a001909. doi: 10.1101/mcs.a001909. Print 2017 Nov. Cold Spring Harb Mol Case Stud. 2017. PMID: 28696212 Free PMC article.
-
Are diagnostic magnetic resonance patterns life-saving in children with biotin-thiamine-responsive basal ganglia disease?Eur J Paediatr Neurol. 2018 Nov;22(6):1139-1149. doi: 10.1016/j.ejpn.2018.06.009. Epub 2018 Jul 9. Eur J Paediatr Neurol. 2018. PMID: 30054086
-
Biotin-thiamine-responsive basal ganglia disease: catastrophic consequences of delay in diagnosis and treatment.Neurol Res. 2017 Feb;39(2):117-125. doi: 10.1080/01616412.2016.1263176. Epub 2016 Dec 1. Neurol Res. 2017. PMID: 27905264
-
Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: a retrospective review of the clinical, radiological and molecular findings of 18 new cases.Orphanet J Rare Dis. 2013 Jun 6;8:83. doi: 10.1186/1750-1172-8-83. Orphanet J Rare Dis. 2013. PMID: 23742248 Free PMC article. Review.
-
Child Neurology: Infantile Biotin Thiamine Responsive Basal Ganglia Disease: Case Report and Brief Review.Neurology. 2023 Apr 25;100(17):836-839. doi: 10.1212/WNL.0000000000206832. Epub 2023 Jan 19. Neurology. 2023. PMID: 36657988 Free PMC article. Review.
Cited by
-
Biotin-thiamine responsive basal ganglia disease: Identification of a pyruvate peak on brain spectroscopy, novel mutation in SLC19A3, and calculation of prevalence based on allele frequencies from aggregated next-generation sequencing data.Am J Med Genet A. 2017 Jun;173(6):1502-1513. doi: 10.1002/ajmg.a.38189. Epub 2017 Apr 12. Am J Med Genet A. 2017. PMID: 28402605 Free PMC article.
-
[Paroxysmal crying and motor regression for more than two months in an infant].Zhongguo Dang Dai Er Ke Za Zhi. 2019 Apr;21(4):399-404. doi: 10.7499/j.issn.1008-8830.2019.04.018. Zhongguo Dang Dai Er Ke Za Zhi. 2019. PMID: 31014436 Free PMC article. Chinese.
-
Compound heterozygous SLC19A3 mutations further refine the critical promoter region for biotin-thiamine-responsive basal ganglia disease.Cold Spring Harb Mol Case Stud. 2017 Nov 21;3(6):a001909. doi: 10.1101/mcs.a001909. Print 2017 Nov. Cold Spring Harb Mol Case Stud. 2017. PMID: 28696212 Free PMC article.
-
Biotin-Thiamine-Responsive Basal Ganglia Disease in Children: A Treatable Neurometabolic Disorder.Ann Indian Acad Neurol. 2021 Mar-Apr;24(2):173-177. doi: 10.4103/aian.AIAN_952_20. Epub 2021 Mar 31. Ann Indian Acad Neurol. 2021. PMID: 34220059 Free PMC article. Review.
-
Potential Role of Genomic Sequencing in the Early Diagnosis of Treatable Genetic Conditions.J Pediatr. 2017 Oct;189:222-226.e1. doi: 10.1016/j.jpeds.2017.06.040. J Pediatr. 2017. PMID: 28947054 Free PMC article.
References
-
- Subramanian VS, Marchant JS, Said HM. Biotin-responsive basal ganglia disease-linked mutations inhibit thiamine transport via hTHTR2: biotin is not a substrate for hTHTR2. American journal of physiology Cell physiology. 2006;291(5):C851–9. - PubMed
-
- Ozand PT, Gascon GG, Al Essa M, Joshi S, Al Jishi E, Bakheet S, et al. Biotin-responsive basal ganglia disease: a novel entity. Brain: a journal of neurology. 1998;121 (Pt 7):1267–79. - PubMed
-
- Tabarki B, Alfadhel M, AlShahwan S, Hundallah K, AlShafi S, AlHashem A. Treatment of biotin-responsive basal ganglia disease: Open comparative study between the combination of biotin plus thiamine versus thiamine alone. European journal of paediatric neurology: EJPN: official journal of the European Paediatric Neurology Society. 2015;19(5):547–52. - PubMed
-
- Tabarki B, Al-Hashem A, Alfadhel M. Biotin-Thiamine-Responsive Basal Ganglia Disease. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R). Seattle (WA)1993. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
