

Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease
- PMID: 26642243
- PMCID: PMC4777523
- DOI: 10.1038/ng.3459
Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease
Abstract
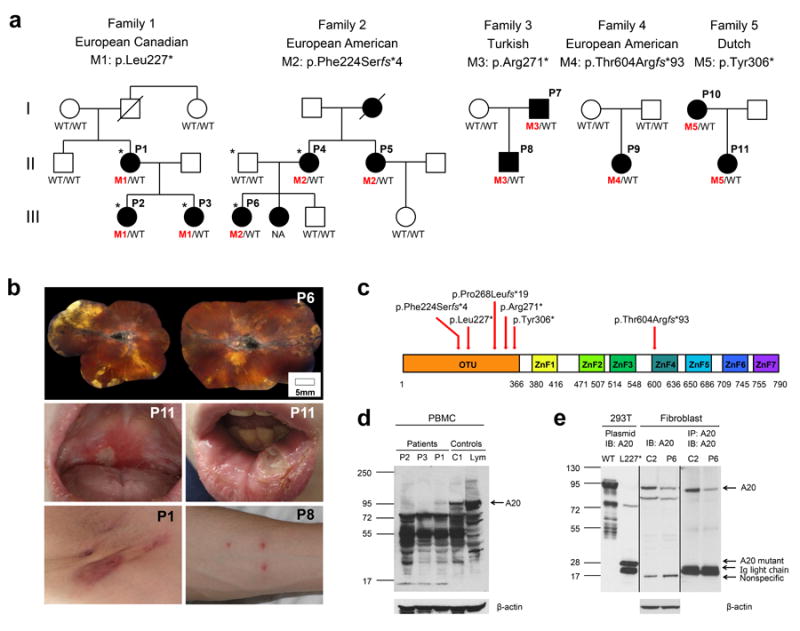
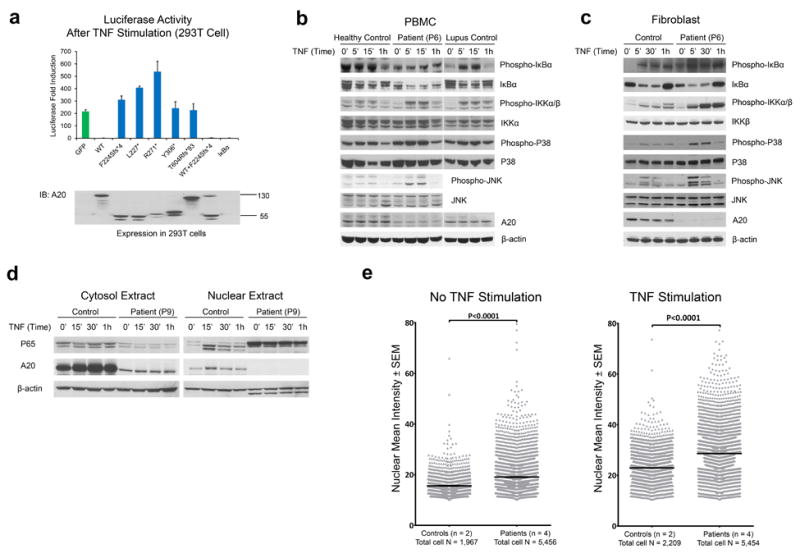
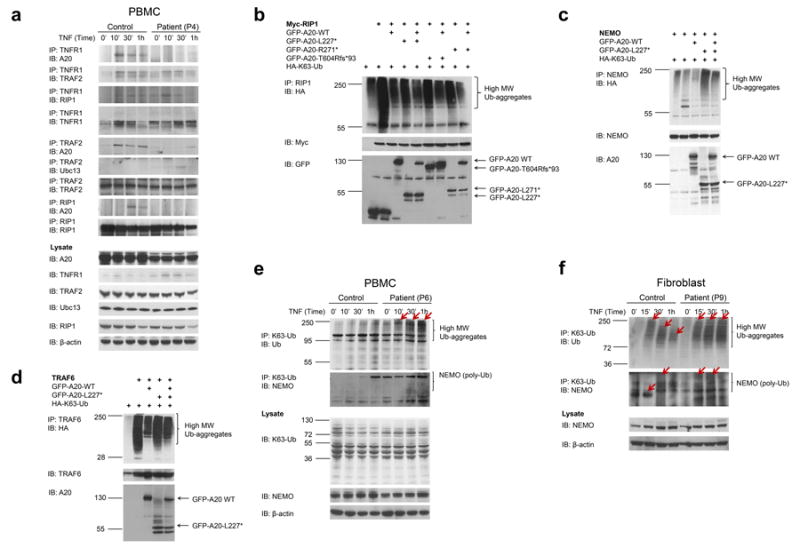
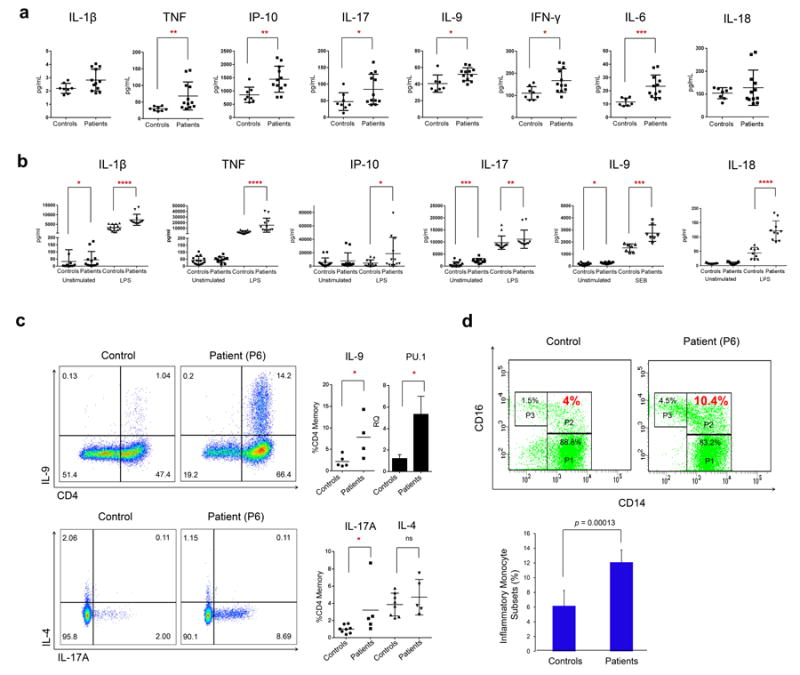
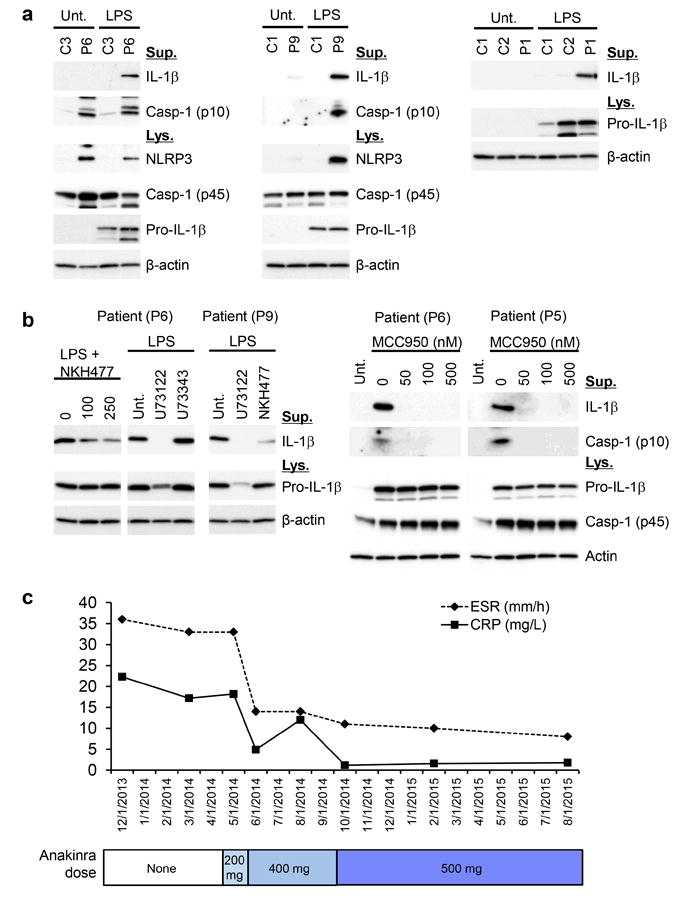
Systemic autoinflammatory diseases are driven by abnormal activation of innate immunity. Herein we describe a new disease caused by high-penetrance heterozygous germline mutations in TNFAIP3, which encodes the NF-κB regulatory protein A20, in six unrelated families with early-onset systemic inflammation. The disorder resembles Behçet's disease, which is typically considered a polygenic disorder with onset in early adulthood. A20 is a potent inhibitor of the NF-κB signaling pathway. Mutant, truncated A20 proteins are likely to act through haploinsufficiency because they do not exert a dominant-negative effect in overexpression experiments. Patient-derived cells show increased degradation of IκBα and nuclear translocation of the NF-κB p65 subunit together with increased expression of NF-κB-mediated proinflammatory cytokines. A20 restricts NF-κB signals via its deubiquitinase activity. In cells expressing mutant A20 protein, there is defective removal of Lys63-linked ubiquitin from TRAF6, NEMO and RIP1 after stimulation with tumor necrosis factor (TNF). NF-κB-dependent proinflammatory cytokines are potential therapeutic targets for the patients with this disease.
Conflict of interest statement
The authors declare no competing financial interests
Figures
Similar articles
-
Recruitment of A20 by the C-terminal domain of NEMO suppresses NF-κB activation and autoinflammatory disease.Proc Natl Acad Sci U S A. 2016 Feb 9;113(6):1612-7. doi: 10.1073/pnas.1518163113. Epub 2016 Jan 22. Proc Natl Acad Sci U S A. 2016. PMID: 26802121 Free PMC article.
-
A20 overexpression inhibits lipopolysaccharide-induced NF-κB activation, TRAF6 and CD40 expression in rat peritoneal mesothelial cells.Int J Mol Sci. 2014 Apr 17;15(4):6592-608. doi: 10.3390/ijms15046592. Int J Mol Sci. 2014. PMID: 24747594 Free PMC article.
-
Downstream activation of NF-κB in the EDA-A1/EDAR signalling in Sjögren's syndrome and its regulation by the ubiquitin-editing enzyme A20.Clin Exp Immunol. 2016 May;184(2):183-96. doi: 10.1111/cei.12764. Epub 2016 Feb 23. Clin Exp Immunol. 2016. PMID: 26724675 Free PMC article.
-
An Update on Autoinflammatory Diseases: Relopathies.Curr Rheumatol Rep. 2018 May 30;20(7):39. doi: 10.1007/s11926-018-0749-x. Curr Rheumatol Rep. 2018. PMID: 29846841 Review.
-
The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology.Trends Immunol. 2009 Aug;30(8):383-91. doi: 10.1016/j.it.2009.05.007. Epub 2009 Jul 28. Trends Immunol. 2009. PMID: 19643665 Review.
Cited by
-
The past 25 years in paediatric rheumatology: insights from monogenic diseases.Nat Rev Rheumatol. 2024 Sep;20(9):585-593. doi: 10.1038/s41584-024-01145-1. Epub 2024 Aug 7. Nat Rev Rheumatol. 2024. PMID: 39112602 Review.
-
Post-translational regulation of inflammasomes.Cell Mol Immunol. 2017 Jan;14(1):65-79. doi: 10.1038/cmi.2016.29. Epub 2016 Jun 27. Cell Mol Immunol. 2017. PMID: 27345727 Free PMC article. Review.
-
Ebosin Ameliorates Psoriasis-Like Inflammation of Mice via miR-155 Targeting tnfaip3 on IL-17 Pathway.Front Immunol. 2021 Apr 26;12:662362. doi: 10.3389/fimmu.2021.662362. eCollection 2021. Front Immunol. 2021. PMID: 33981308 Free PMC article.
-
The PANoptosome: A Deadly Protein Complex Driving Pyroptosis, Apoptosis, and Necroptosis (PANoptosis).Front Cell Infect Microbiol. 2020 Jun 3;10:238. doi: 10.3389/fcimb.2020.00238. eCollection 2020. Front Cell Infect Microbiol. 2020. PMID: 32582562 Free PMC article. Review.
-
"Deficiency in ELF4, X-Linked": a Monogenic Disease Entity Resembling Behçet's Syndrome and Inflammatory Bowel Disease.J Clin Immunol. 2024 Jan 17;44(2):44. doi: 10.1007/s10875-023-01610-8. J Clin Immunol. 2024. PMID: 38231408 Free PMC article.
References
-
- van Kempen TS, Wenink MH, Leijten EF, Radstake TR, Boes M. Perception of self: distinguishing autoimmunity from autoinflammation. Nature reviews Rheumatology. 2015 - PubMed
-
- Sakane T, Takeno M, Suzuki N, Inaba G. Behcet’s disease. The New England journal of medicine. 1999;341:1284–91. - PubMed
-
- Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends in immunology. 2014;35:22–31. - PubMed
-
- Russo RA, Brogan PA. Monogenic autoinflammatory diseases. Rheumatology. 2014;53:1927–39. - PubMed
-
- Martinon F, Aksentijevich I. New players driving inflammation in monogenic autoinflammatory diseases. Nature reviews. Rheumatology. 2015;11:11– 20. - PubMed
Methods-only references
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
