

Mutations Impairing GSK3-Mediated MAF Phosphorylation Cause Cataract, Deafness, Intellectual Disability, Seizures, and a Down Syndrome-like Facies
- PMID: 25865493
- PMCID: PMC4570552
- DOI: 10.1016/j.ajhg.2015.03.001
Mutations Impairing GSK3-Mediated MAF Phosphorylation Cause Cataract, Deafness, Intellectual Disability, Seizures, and a Down Syndrome-like Facies
Abstract
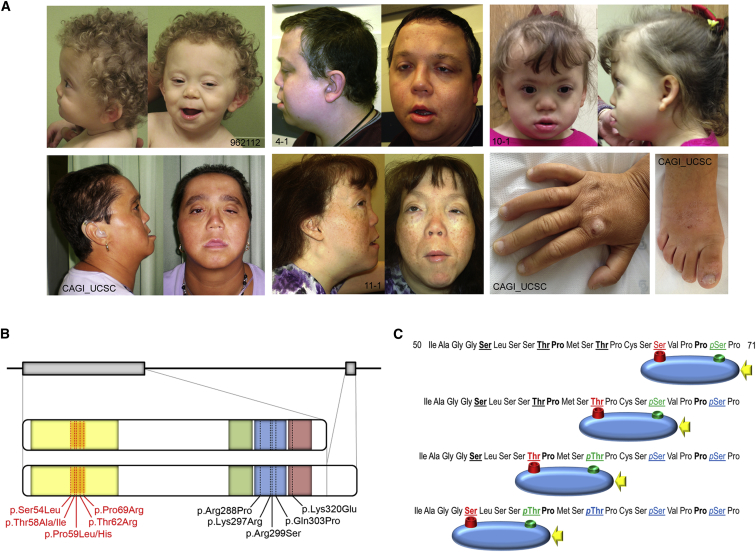
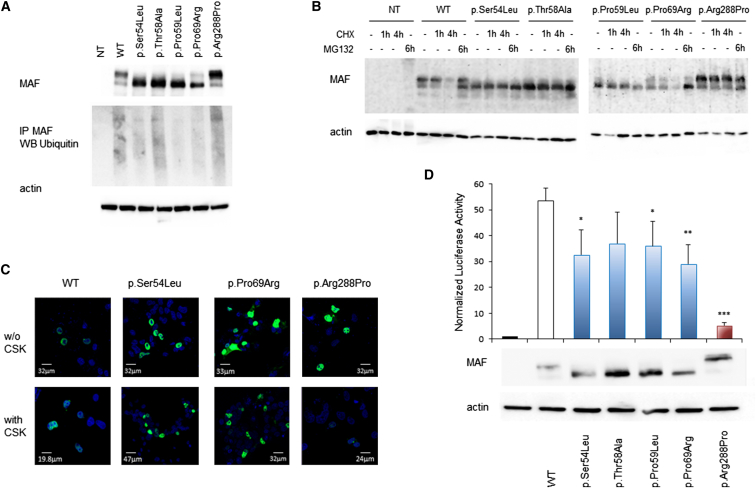
Transcription factors operate in developmental processes to mediate inductive events and cell competence, and perturbation of their function or regulation can dramatically affect morphogenesis, organogenesis, and growth. We report that a narrow spectrum of amino-acid substitutions within the transactivation domain of the v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog (MAF), a leucine zipper-containing transcription factor of the AP1 superfamily, profoundly affect development. Seven different de novo missense mutations involving conserved residues of the four GSK3 phosphorylation motifs were identified in eight unrelated individuals. The distinctive clinical phenotype, for which we propose the eponym Aymé-Gripp syndrome, is not limited to lens and eye defects as previously reported for MAF/Maf loss of function but includes sensorineural deafness, intellectual disability, seizures, brachycephaly, distinctive flat facial appearance, skeletal anomalies, mammary gland hypoplasia, and reduced growth. Disease-causing mutations were demonstrated to impair proper MAF phosphorylation, ubiquitination and proteasomal degradation, perturbed gene expression in primary skin fibroblasts, and induced neurodevelopmental defects in an in vivo model. Our findings nosologically and clinically delineate a previously poorly understood recognizable multisystem disorder, provide evidence for MAF governing a wider range of developmental programs than previously appreciated, and describe a novel instance of protein dosage effect severely perturbing development.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures


Similar articles
-
Further delineation of Aymé-Gripp syndrome and use of automated facial analysis tool.Am J Med Genet A. 2018 Jul;176(7):1648-1656. doi: 10.1002/ajmg.a.38832. Am J Med Genet A. 2018. PMID: 30160832
-
Skeletal abnormalities are common features in Aymé-Gripp syndrome.Clin Genet. 2020 Feb;97(2):362-369. doi: 10.1111/cge.13651. Epub 2019 Nov 3. Clin Genet. 2020. PMID: 31600839
-
Ayme gripp syndrome in an Indian patient.Am J Med Genet A. 2021 Apr;185(4):1312-1316. doi: 10.1002/ajmg.a.62053. Epub 2021 Jan 1. Am J Med Genet A. 2021. PMID: 33528093
-
Mutation update of transcription factor genes FOXE3, HSF4, MAF, and PITX3 causing cataracts and other developmental ocular defects.Hum Mutat. 2018 Apr;39(4):471-494. doi: 10.1002/humu.23395. Epub 2018 Jan 16. Hum Mutat. 2018. PMID: 29314435 Free PMC article. Review.
-
Identification of a novel missense mutation of MAF in a Japanese family with congenital cataract by whole exome sequencing: a clinical report and review of literature.Am J Med Genet A. 2014 May;164A(5):1272-6. doi: 10.1002/ajmg.a.36433. Epub 2014 Mar 24. Am J Med Genet A. 2014. PMID: 24664492 Review.
Cited by
-
Glycogen Synthase Kinase-3 Inhibitors: Preclinical and Clinical Focus on CNS-A Decade Onward.Front Mol Neurosci. 2022 Jan 21;14:792364. doi: 10.3389/fnmol.2021.792364. eCollection 2021. Front Mol Neurosci. 2022. PMID: 35126052 Free PMC article. Review.
-
Role of MafB in macrophages.Exp Anim. 2020 Jan 29;69(1):1-10. doi: 10.1538/expanim.19-0076. Epub 2019 Oct 1. Exp Anim. 2020. PMID: 31582643 Free PMC article. Review.
-
The role of post-translational modifications in hearing and deafness.Cell Mol Life Sci. 2016 Sep;73(18):3521-33. doi: 10.1007/s00018-016-2257-3. Epub 2016 May 4. Cell Mol Life Sci. 2016. PMID: 27147466 Free PMC article. Review.
-
Second MAFA Variant Causing a Phosphorylation Defect in the Transactivation Domain and Familial Insulinomatosis.Cancers (Basel). 2022 Apr 1;14(7):1798. doi: 10.3390/cancers14071798. Cancers (Basel). 2022. PMID: 35406570 Free PMC article.
-
Exploring Large MAF Transcription Factors: Functions, Pathology, and Mouse Models with Point Mutations.Genes (Basel). 2023 Sep 27;14(10):1883. doi: 10.3390/genes14101883. Genes (Basel). 2023. PMID: 37895232 Free PMC article. Review.
References
-
- Gripp K.W., Nicholson L., Scott C.I., Jr. Apparently new syndrome of congenital cataracts, sensorineural deafness, Down syndrome-like facial appearance, short stature, and mental retardation. Am. J. Med. Genet. 1996;61:382–386. - PubMed
-
- Aymé S., Philip N. Fine-Lubinsky syndrome: a fourth patient with brachycephaly, deafness, cataract, microstomia and mental retardation. Clin. Dysmorphol. 1996;5:55–60. - PubMed
-
- Fine B.A., Lubinsky M. Craniofacial and CNS anomalies with body asymmetry, severe retardation, and other malformations. J. Clin. Dysmorphol. 1983;1:6–9. - PubMed
-
- Aymé S., Philip N. Apparently new syndrome of congenital cataracts, sensorineural deafness, Down syndrome-like facial appearance, short stature, and mental retardation. Am. J. Med. Genet. 1997;70:333–335. - PubMed
-
- Keppler-Noreuil K., Welch J., Baker-Lange K. Syndrome of congenital cataracts, sensorineural deafness, Down syndrome-like facial appearance, short stature, and mental retardation: two additional cases. Am. J. Med. Genet. A. 2007;143A:2581–2587. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
