

Germline recessive mutations in PI4KA are associated with perisylvian polymicrogyria, cerebellar hypoplasia and arthrogryposis
- PMID: 25855803
- PMCID: PMC4459391
- DOI: 10.1093/hmg/ddv117
Germline recessive mutations in PI4KA are associated with perisylvian polymicrogyria, cerebellar hypoplasia and arthrogryposis
Abstract
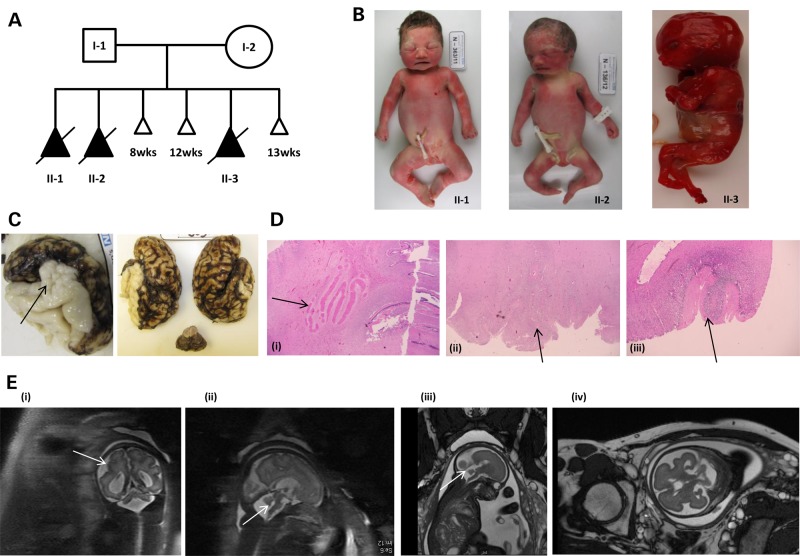
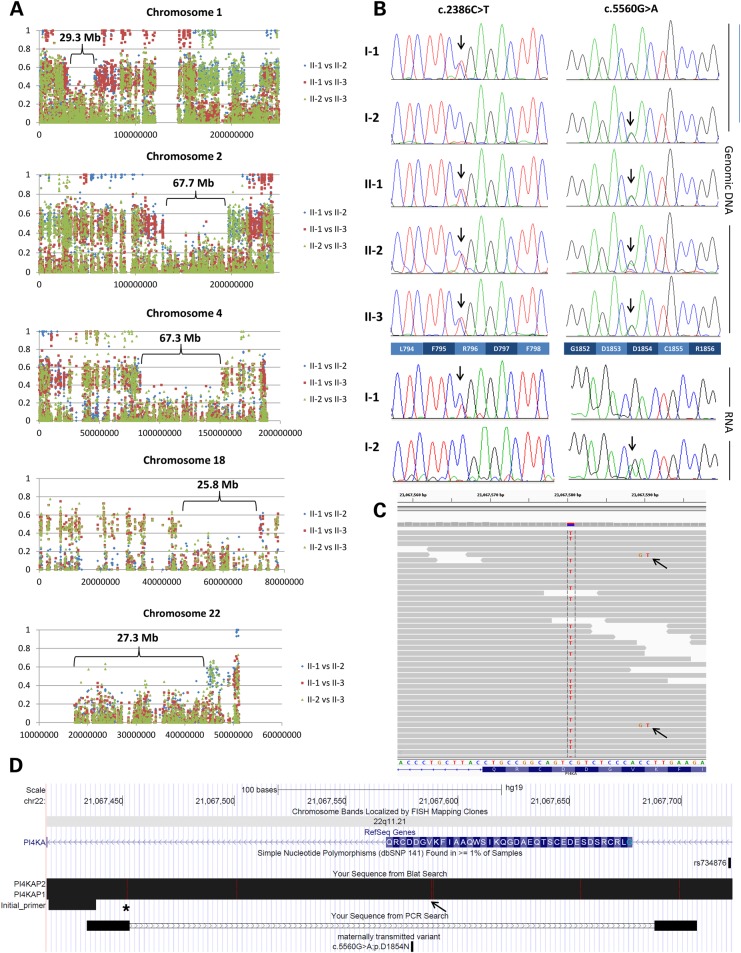
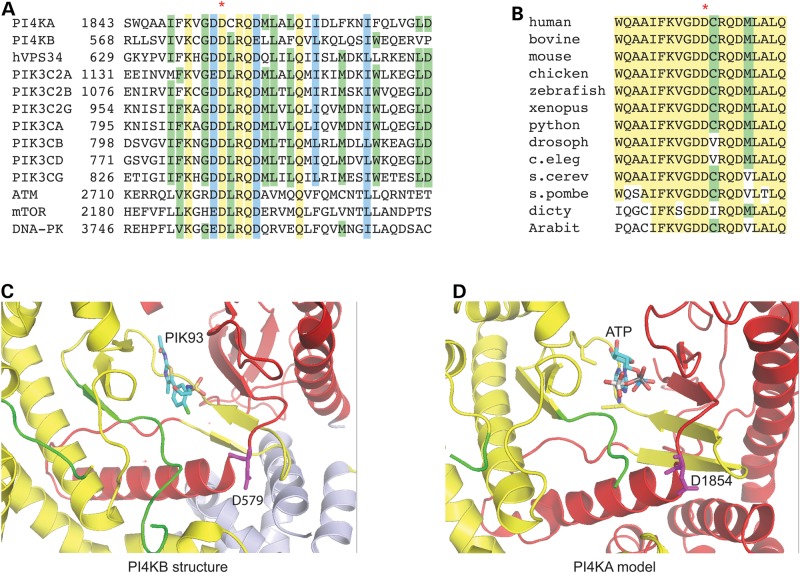
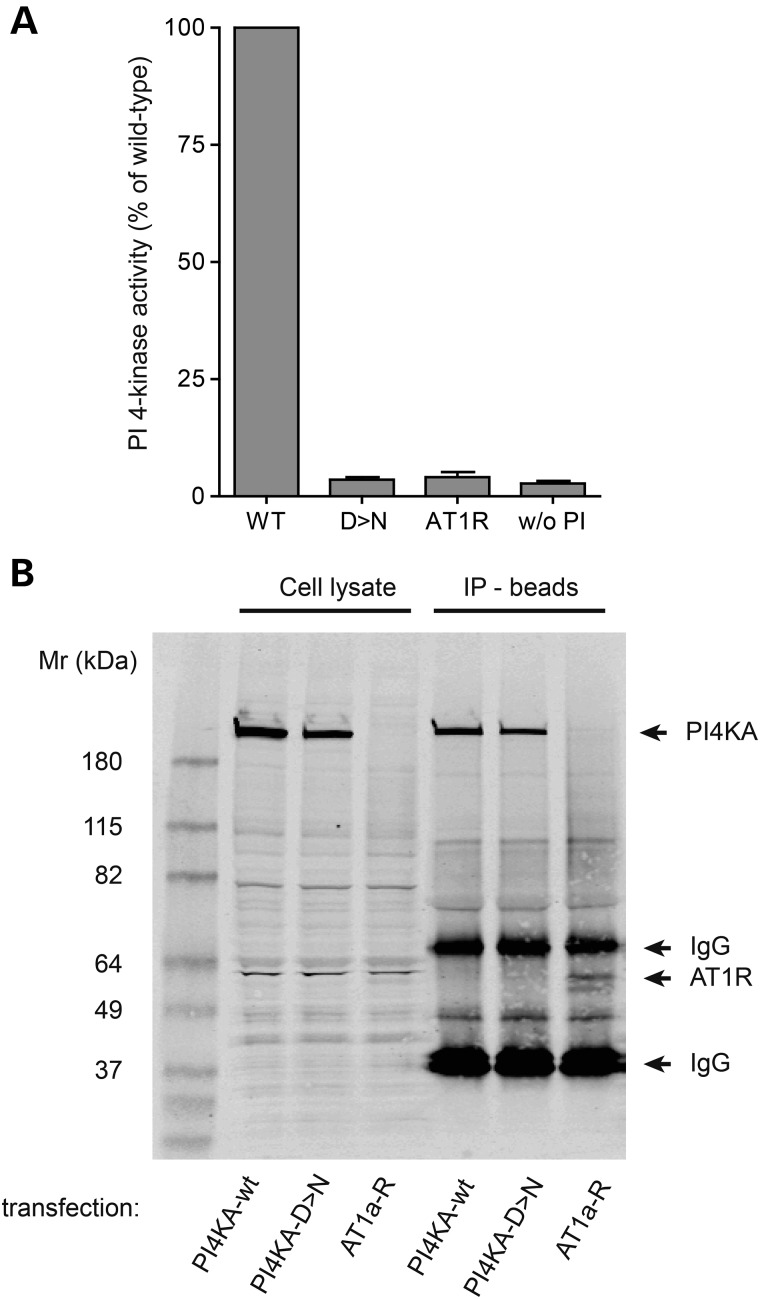
Polymicrogyria (PMG) is a structural brain abnormality involving the cerebral cortex that results from impaired neuronal migration and although several genes have been implicated, many cases remain unsolved. In this study, exome sequencing in a family where three fetuses had all been diagnosed with PMG and cerebellar hypoplasia allowed us to identify regions of the genome for which both chromosomes were shared identical-by-descent, reducing the search space for causative variants to 8.6% of the genome. In these regions, the only plausibly pathogenic mutations were compound heterozygous variants in PI4KA, which Sanger sequencing confirmed segregated consistent with autosomal recessive inheritance. The paternally transmitted variant predicted a premature stop mutation (c.2386C>T; p.R796X), whereas the maternally transmitted variant predicted a missense substitution (c.5560G>A; p.D1854N) at a conserved residue within the catalytic domain. Functional studies using expressed wild-type or mutant PI4KA enzyme confirmed the importance of p.D1854 for kinase activity. Our results emphasize the importance of phosphoinositide signalling in early brain development.
© The Author 2015. Published by Oxford University Press.
Figures
Similar articles
-
Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with a loss-of-function mutation in CDK5.Hum Genet. 2015 Mar;134(3):305-14. doi: 10.1007/s00439-014-1522-5. Epub 2015 Jan 6. Hum Genet. 2015. PMID: 25560765
-
WDR73 missense mutation causes infantile onset intellectual disability and cerebellar hypoplasia in a consanguineous family.Clin Chim Acta. 2017 Jan;464:24-29. doi: 10.1016/j.cca.2016.10.029. Epub 2016 Oct 28. Clin Chim Acta. 2017. PMID: 27983999
-
A missense variant in ITPR1 provides evidence for autosomal recessive SCA29 with asymptomatic cerebellar hypoplasia in carriers.Eur J Hum Genet. 2017 Jun;25(7):848-853. doi: 10.1038/ejhg.2017.54. Epub 2017 May 10. Eur J Hum Genet. 2017. PMID: 28488678 Free PMC article.
-
Epileptogenic brain malformations: clinical presentation, malformative patterns and indications for genetic testing.Seizure. 2002 Apr;11 Suppl A:532-43; quiz 544-7. Seizure. 2002. PMID: 12185771 Review.
-
Genetic malformations of the cerebral cortex and epilepsy.Epilepsia. 2005;46 Suppl 1:32-7. doi: 10.1111/j.0013-9580.2005.461010.x. Epilepsia. 2005. PMID: 15816977 Review.
Cited by
-
Phosphoinositides signaling modulates microglial actin remodeling and phagocytosis in Alzheimer's disease.Cell Commun Signal. 2021 Feb 24;19(1):28. doi: 10.1186/s12964-021-00715-0. Cell Commun Signal. 2021. PMID: 33627135 Free PMC article. Review.
-
Beyond PI3Ks: targeting phosphoinositide kinases in disease.Nat Rev Drug Discov. 2023 May;22(5):357-386. doi: 10.1038/s41573-022-00582-5. Epub 2022 Nov 14. Nat Rev Drug Discov. 2023. PMID: 36376561 Free PMC article. Review.
-
New Cohort of Patients With CEDNIK Syndrome Expands the Phenotypic and Genotypic Spectra.Neurol Genet. 2021 Jan 12;7(1):e553. doi: 10.1212/NXG.0000000000000553. eCollection 2021 Feb. Neurol Genet. 2021. PMID: 33977139 Free PMC article.
-
A synonymous mutation in PI4KA impacts the transcription and translation process of gene expression.Front Immunol. 2022 Oct 19;13:987666. doi: 10.3389/fimmu.2022.987666. eCollection 2022. Front Immunol. 2022. PMID: 36341355 Free PMC article.
-
Chromothripsis and ring chromosome 22: a paradigm of genomic complexity in the Phelan-McDermid syndrome (22q13 deletion syndrome).J Med Genet. 2018 Apr;55(4):269-277. doi: 10.1136/jmedgenet-2017-105125. Epub 2018 Jan 29. J Med Genet. 2018. PMID: 29378768 Free PMC article.
References
-
- Levine D.N., Fisher M.A., Caviness V.S., Jr (1974) Porencephaly with microgyria: a pathologic study. Acta Neuropathol., 29, 99–113. - PubMed
-
- Chang B.S., Piao X., Bodell A., Basel-Vanagaite L., Straussberg R., Dobyns W.B., Qasrawi B., Winter R.M., Innes A.M., Voit T., et al. (2003) Bilateral frontoparietal polymicrogyria: clinical and radiological features in 10 families with linkage to chromosome 16. Ann. Neurol., 53, 596–606. - PubMed
-
- Guerreiro M.M., Andermann E., Guerrini R., Dobyns W.B., Kuzniecky R., Silver K., Van Bogaert P., Gillain C., David P., Ambrosetto G., et al. (2000) Familial perisylvian polymicrogyria: a new familial syndrome of cortical maldevelopment. Ann. Neurol., 48, 39–48. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
