

Structural modeling of tissue-specific mitochondrial alanyl-tRNA synthetase (AARS2) defects predicts differential effects on aminoacylation
- PMID: 25705216
- PMCID: PMC4319469
- DOI: 10.3389/fgene.2015.00021
Structural modeling of tissue-specific mitochondrial alanyl-tRNA synthetase (AARS2) defects predicts differential effects on aminoacylation
Abstract
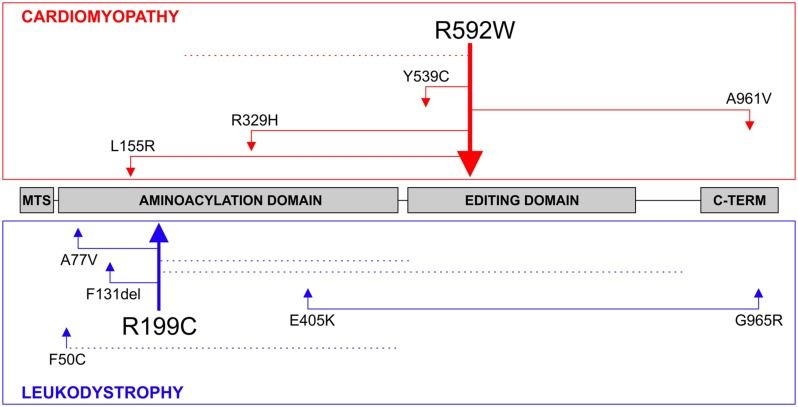
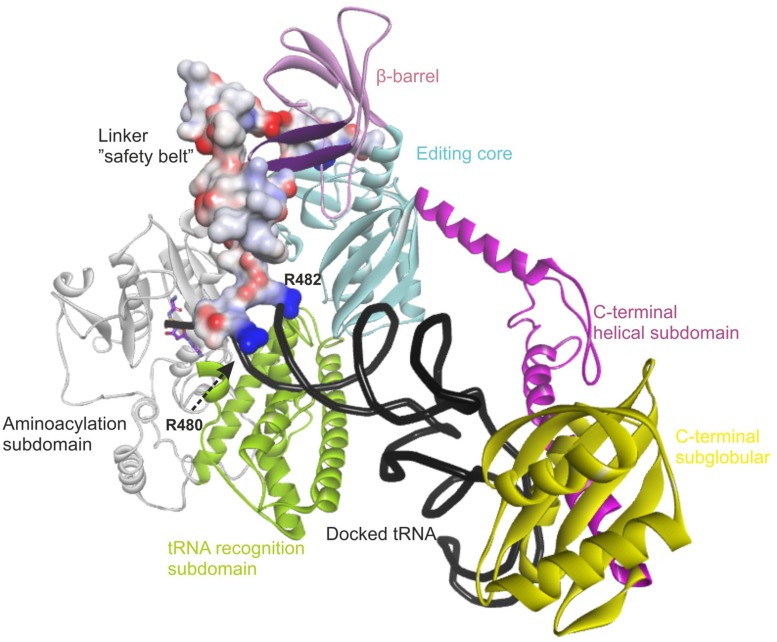
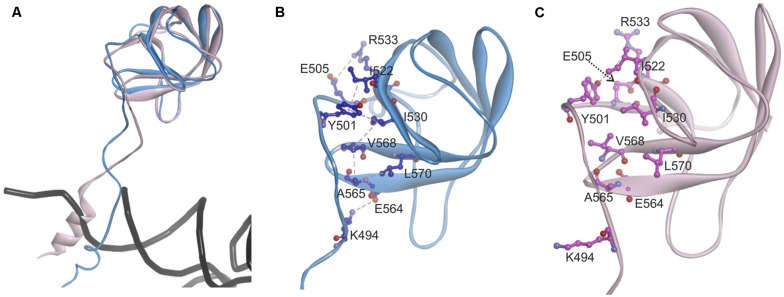
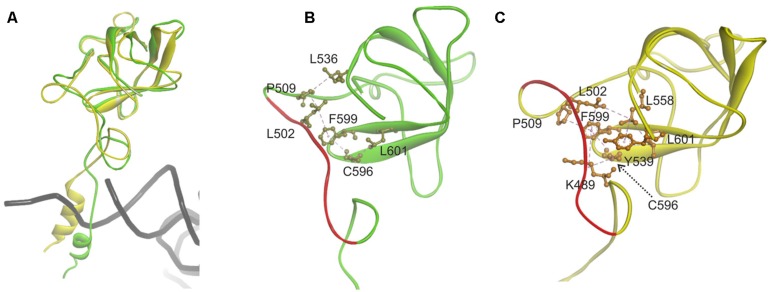
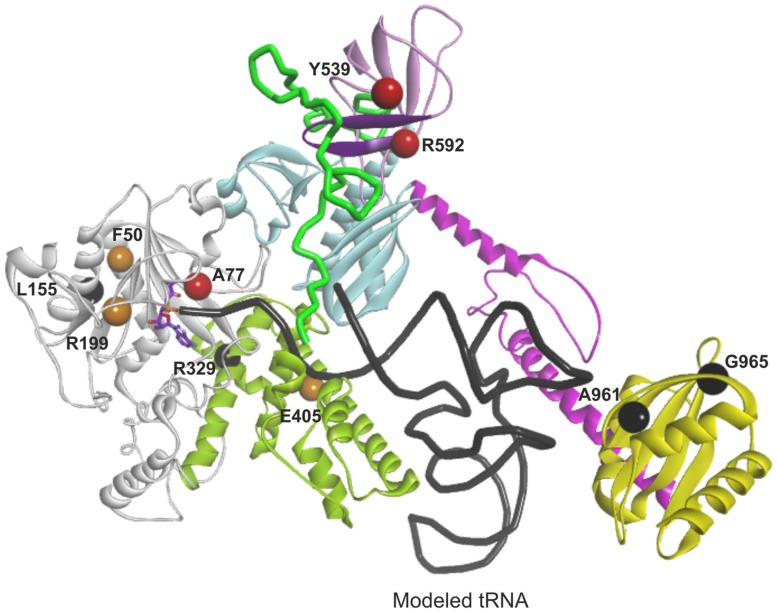
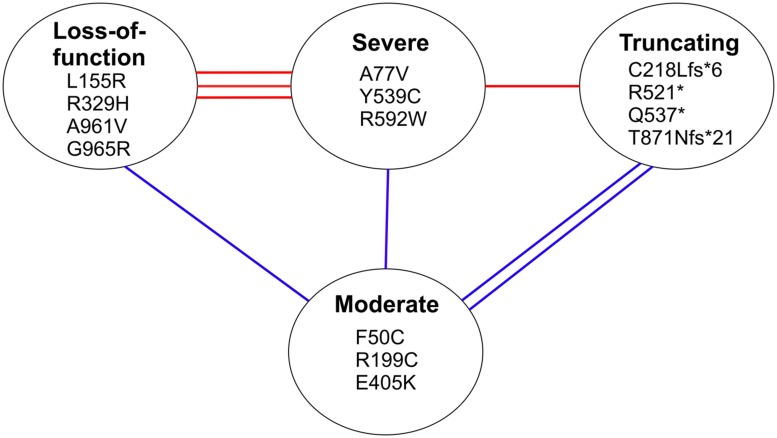
The accuracy of mitochondrial protein synthesis is dependent on the coordinated action of nuclear-encoded mitochondrial aminoacyl-tRNA synthetases (mtARSs) and the mitochondrial DNA-encoded tRNAs. The recent advances in whole-exome sequencing have revealed the importance of the mtARS proteins for mitochondrial pathophysiology since nearly every nuclear gene for mtARS (out of 19) is now recognized as a disease gene for mitochondrial disease. Typically, defects in each mtARS have been identified in one tissue-specific disease, most commonly affecting the brain, or in one syndrome. However, mutations in the AARS2 gene for mitochondrial alanyl-tRNA synthetase (mtAlaRS) have been reported both in patients with infantile-onset cardiomyopathy and in patients with childhood to adulthood-onset leukoencephalopathy. We present here an investigation of the effects of the described mutations on the structure of the synthetase, in an effort to understand the tissue-specific outcomes of the different mutations. The mtAlaRS differs from the other mtARSs because in addition to the aminoacylation domain, it has a conserved editing domain for deacylating tRNAs that have been mischarged with incorrect amino acids. We show that the cardiomyopathy phenotype results from a single allele, causing an amino acid change R592W in the editing domain of AARS2, whereas the leukodystrophy mutations are located in other domains of the synthetase. Nevertheless, our structural analysis predicts that all mutations reduce the aminoacylation activity of the synthetase, because all mtAlaRS domains contribute to tRNA binding for aminoacylation. According to our model, the cardiomyopathy mutations severely compromise aminoacylation whereas partial activity is retained by the mutation combinations found in the leukodystrophy patients. These predictions provide a hypothesis for the molecular basis of the distinct tissue-specific phenotypic outcomes.
Keywords: alanyl-tRNA synthetase; aminoacyl-tRNA synthetases; mitochondrial disease; structural modeling; tissue-specificity.
Figures

Similar articles
-
Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy.Am J Hum Genet. 2011 May 13;88(5):635-42. doi: 10.1016/j.ajhg.2011.04.006. Epub 2011 May 5. Am J Hum Genet. 2011. PMID: 21549344 Free PMC article.
-
Novel alanyl-tRNA synthetase 2 (AARS2) homozygous mutation in a consanguineous Chinese family with premature ovarian insufficiency.Fertil Steril. 2019 Sep;112(3):569-576.e2. doi: 10.1016/j.fertnstert.2019.05.005. Epub 2019 Jul 4. Fertil Steril. 2019. PMID: 31280959
-
Mitochondrial aminoacyl-tRNA synthetase disorders: an emerging group of developmental disorders of myelination.J Neurodev Disord. 2019 Dec 16;11(1):29. doi: 10.1186/s11689-019-9292-y. J Neurodev Disord. 2019. PMID: 31839000 Free PMC article. Review.
-
Case report: 'AARS2 leukodystrophy'.Mol Genet Metab Rep. 2021 Jul 13;28:100782. doi: 10.1016/j.ymgmr.2021.100782. eCollection 2021 Sep. Mol Genet Metab Rep. 2021. PMID: 34285876 Free PMC article.
-
Trans-editing by aminoacyl-tRNA synthetase-like editing domains.Enzymes. 2020;48:69-115. doi: 10.1016/bs.enz.2020.07.002. Epub 2020 Sep 8. Enzymes. 2020. PMID: 33837712 Review.
Cited by
-
Loss-of-function mutations in Lysyl-tRNA synthetase cause various leukoencephalopathy phenotypes.Neurol Genet. 2019 Apr 18;5(2):e565. doi: 10.1212/NXG.0000000000000316. eCollection 2019 Apr. Neurol Genet. 2019. PMID: 31192300 Free PMC article.
-
The first Japanese case of leukodystrophy with ovarian failure arising from novel compound heterozygous AARS2 mutations.J Hum Genet. 2016 Oct;61(10):899-902. doi: 10.1038/jhg.2016.64. Epub 2016 Jun 2. J Hum Genet. 2016. PMID: 27251004
-
Novel AARS2 gene mutation producing leukodystrophy: a case report.J Hum Genet. 2017 Feb;62(2):329-333. doi: 10.1038/jhg.2016.126. Epub 2016 Oct 13. J Hum Genet. 2017. PMID: 27734837
-
Mitochondrial Aminoacyl-tRNA Synthetase and Disease: The Yeast Contribution for Functional Analysis of Novel Variants.Int J Mol Sci. 2021 Apr 26;22(9):4524. doi: 10.3390/ijms22094524. Int J Mol Sci. 2021. PMID: 33926074 Free PMC article. Review.
-
The emerging neurological spectrum of AARS2-associated disorders.Parkinsonism Relat Disord. 2021 Dec;93:50-54. doi: 10.1016/j.parkreldis.2021.10.031. Epub 2021 Nov 10. Parkinsonism Relat Disord. 2021. PMID: 34784527 Free PMC article. Review.
References
-
- Bayat V., Thiffault I., Jaiswal M., Tetreault M., Donti T., Sasarman F., et al. (2012). Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 10:e1001288 10.1371/journal.pbio.1001288 - DOI - PMC - PubMed
-
- Belostotsky R., Ben-Shalom E., Rinat C., Becker-Cohen R., Feinstein S., Zeligson S., et al. (2011). Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am. J. Hum. Genet. 88 193–200 10.1016/j.ajhg.2010.12.010 - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources