

Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data
- PMID: 25529582
- PMCID: PMC4392068
- DOI: 10.1016/S0140-6736(14)61705-0
Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data
Abstract
Background: Human genome sequencing has transformed our understanding of genomic variation and its relevance to health and disease, and is now starting to enter clinical practice for the diagnosis of rare diseases. The question of whether and how some categories of genomic findings should be shared with individual research participants is currently a topic of international debate, and development of robust analytical workflows to identify and communicate clinically relevant variants is paramount.
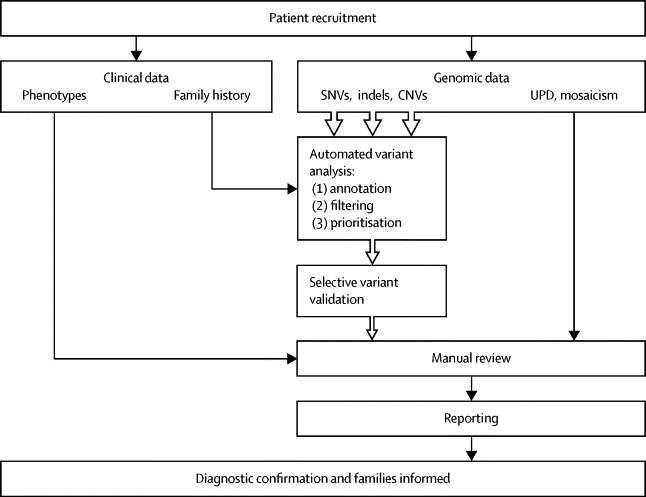
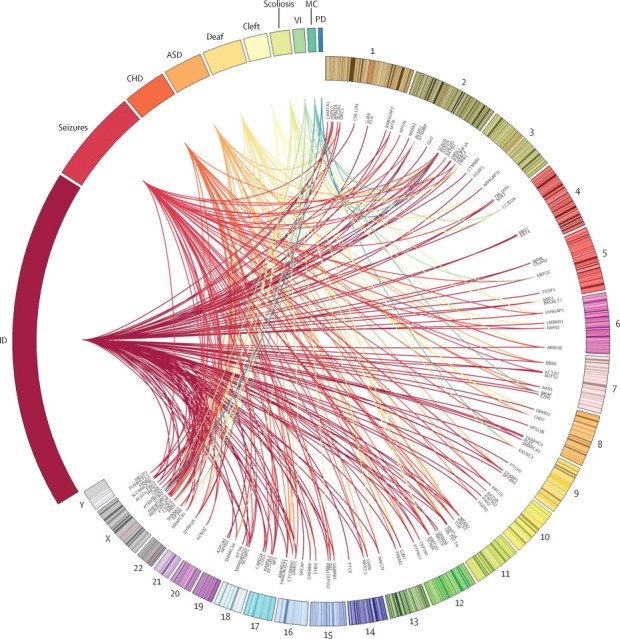
Methods: The Deciphering Developmental Disorders (DDD) study has developed a UK-wide patient recruitment network involving over 180 clinicians across all 24 regional genetics services, and has performed genome-wide microarray and whole exome sequencing on children with undiagnosed developmental disorders and their parents. After data analysis, pertinent genomic variants were returned to individual research participants via their local clinical genetics team.
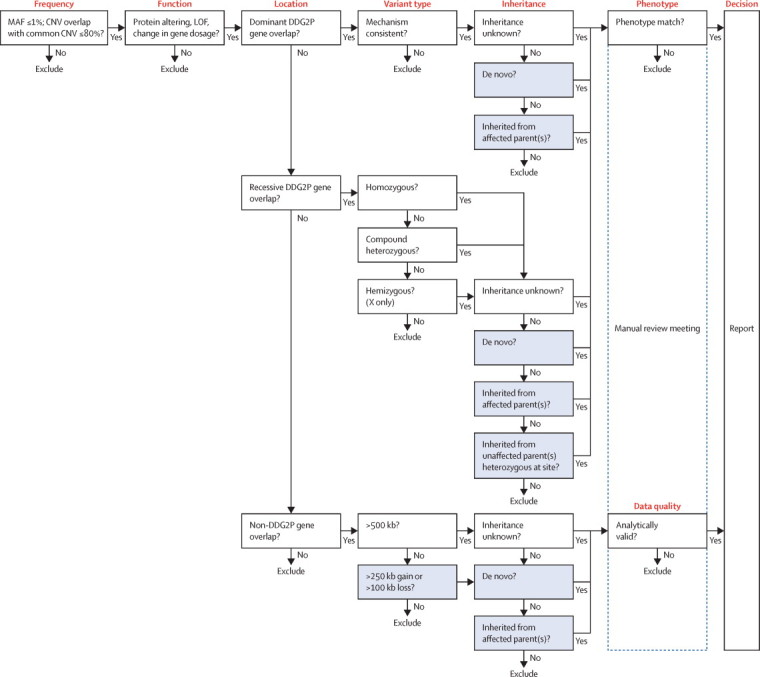
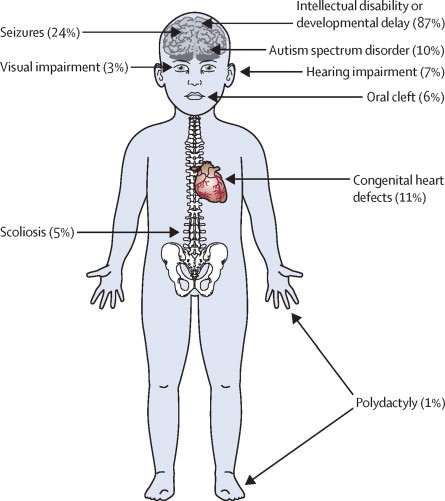
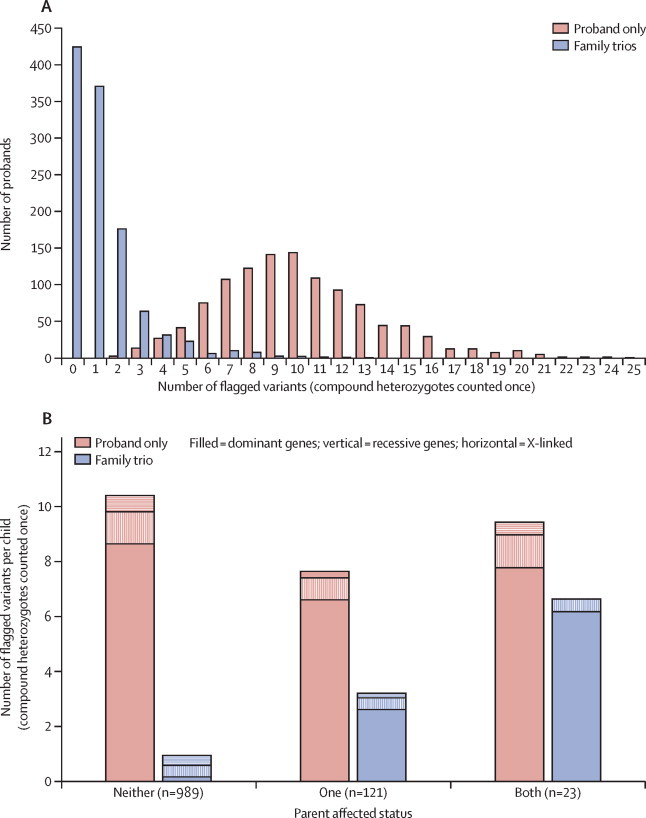
Findings: Around 80,000 genomic variants were identified from exome sequencing and microarray analysis in each individual, of which on average 400 were rare and predicted to be protein altering. By focusing only on de novo and segregating variants in known developmental disorder genes, we achieved a diagnostic yield of 27% among 1133 previously investigated yet undiagnosed children with developmental disorders, whilst minimising incidental findings. In families with developmentally normal parents, whole exome sequencing of the child and both parents resulted in a 10-fold reduction in the number of potential causal variants that needed clinical evaluation compared to sequencing only the child. Most diagnostic variants identified in known genes were novel and not present in current databases of known disease variation.
Interpretation: Implementation of a robust translational genomics workflow is achievable within a large-scale rare disease research study to allow feedback of potentially diagnostic findings to clinicians and research participants. Systematic recording of relevant clinical data, curation of a gene-phenotype knowledge base, and development of clinical decision support software are needed in addition to automated exclusion of almost all variants, which is crucial for scalable prioritisation and review of possible diagnostic variants. However, the resource requirements of development and maintenance of a clinical reporting system within a research setting are substantial.
Funding: Health Innovation Challenge Fund, a parallel funding partnership between the Wellcome Trust and the UK Department of Health.
Copyright © 2015 Wright et al. Open Access article distributed under the terms of CC BY. Published by Elsevier Ltd. All rights reserved.
Figures
Comment in
-
Developmental disorders: deciphering exomes on a grand scale.Lancet. 2015 Apr 4;385(9975):1266-7. doi: 10.1016/S0140-6736(14)62122-X. Epub 2014 Dec 17. Lancet. 2015. PMID: 25529583 No abstract available.
Similar articles
-
Monogenic variants in dystonia: an exome-wide sequencing study.Lancet Neurol. 2020 Nov;19(11):908-918. doi: 10.1016/S1474-4422(20)30312-4. Lancet Neurol. 2020. PMID: 33098801 Free PMC article.
-
A Cross-Disorder Method to Identify Novel Candidate Genes for Developmental Brain Disorders.JAMA Psychiatry. 2016 Mar;73(3):275-83. doi: 10.1001/jamapsychiatry.2015.2692. JAMA Psychiatry. 2016. PMID: 26817790 Free PMC article.
-
Genome-Wide Sequencing for Unexplained Developmental Disabilities or Multiple Congenital Anomalies: A Health Technology Assessment.Ont Health Technol Assess Ser. 2020 Mar 6;20(11):1-178. eCollection 2020. Ont Health Technol Assess Ser. 2020. PMID: 32194879 Free PMC article.
-
Whole exome or genome sequencing: nurses need to prepare families for the possibilities.J Adv Nurs. 2014 Dec;70(12):2736-45. doi: 10.1111/jan.12516. Epub 2014 Sep 1. J Adv Nurs. 2014. PMID: 25175401 Review.
-
DECIPHER: web-based, community resource for clinical interpretation of rare variants in developmental disorders.Hum Mol Genet. 2012 Oct 15;21(R1):R37-44. doi: 10.1093/hmg/dds362. Epub 2012 Sep 8. Hum Mol Genet. 2012. PMID: 22962312 Free PMC article. Review.
Cited by
-
Biallelic variants in DAP3 result in reduced assembly of the mitoribosomal small subunit with altered intrinsic and extrinsic apoptosis and a Perrault syndrome-spectrum phenotype.medRxiv [Preprint]. 2024 Aug 21:2024.08.19.24312079. doi: 10.1101/2024.08.19.24312079. medRxiv. 2024. Update in: Am J Hum Genet. 2025 Jan 2;112(1):59-74. doi: 10.1016/j.ajhg.2024.11.007. PMID: 39371131 Free PMC article. Updated. Preprint.
-
Exon-focused targeted oligonucleotide microarray design increases detection of clinically relevant variants across multiple NHS genomic centres.NPJ Genom Med. 2020 Jul 21;5:28. doi: 10.1038/s41525-020-0136-1. eCollection 2020. NPJ Genom Med. 2020. PMID: 32714564 Free PMC article.
-
CNV analysis in Chinese children of mental retardation highlights a sex differentiation in parental contribution to de novo and inherited mutational burdens.Sci Rep. 2016 Jun 3;6:25954. doi: 10.1038/srep25954. Sci Rep. 2016. PMID: 27257017 Free PMC article.
-
Review: Cancer and neurodevelopmental disorders: multi-scale reasoning and computational guide.Front Cell Dev Biol. 2024 Jul 2;12:1376639. doi: 10.3389/fcell.2024.1376639. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 39015651 Free PMC article. Review.
-
De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability.Am J Med Genet A. 2015 Oct;167A(10):2231-7. doi: 10.1002/ajmg.a.37189. Epub 2015 Jun 15. Am J Med Genet A. 2015. PMID: 26079862 Free PMC article.
References
-
- Bamshad MJ, Ng SB, Bigham AW. Exome sequencing as a tool for mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. - PubMed
-
- Worthey EA, Mayer AN, Syverson GD. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–262. - PubMed
-
- de Ligt J, Willemsen MH, van Bon BWM. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–1929. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
