

Congenital hypogonadotropic hypogonadism with split hand/foot malformation: a clinical entity with a high frequency of FGFR1 mutations
- PMID: 25394172
- PMCID: PMC4430466
- DOI: 10.1038/gim.2014.166
Congenital hypogonadotropic hypogonadism with split hand/foot malformation: a clinical entity with a high frequency of FGFR1 mutations
Abstract
Purpose: Congenital hypogonadotropic hypogonadism (CHH) and split hand/foot malformation (SHFM) are two rare genetic conditions. Here we report a clinical entity comprising the two.
Methods: We identified patients with CHH and SHFM through international collaboration. Probands and available family members underwent phenotyping and screening for FGFR1 mutations. The impact of identified mutations was assessed by sequence- and structure-based predictions and/or functional assays.
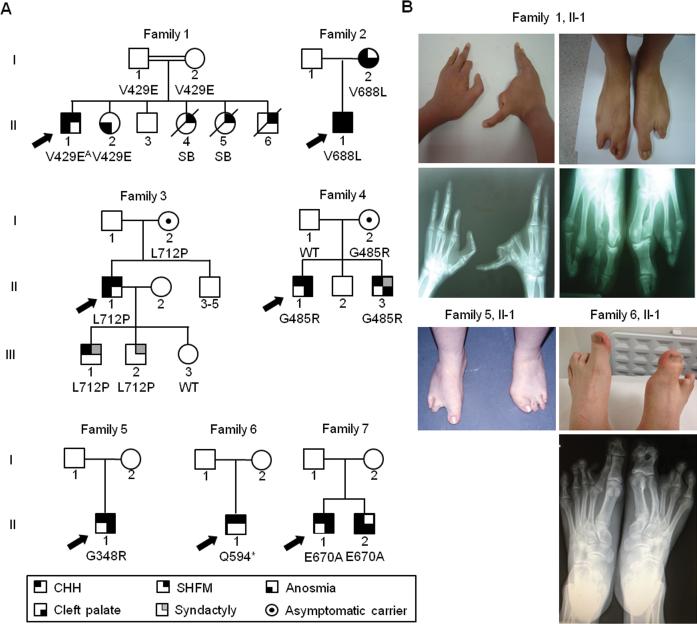
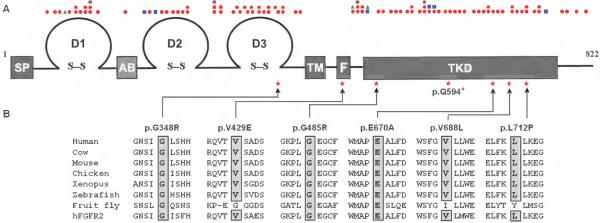
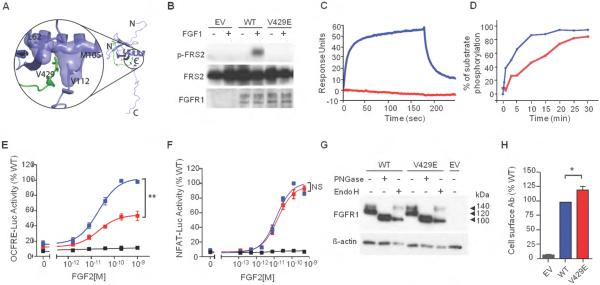
Results: We identified eight probands with CHH with (n = 3; Kallmann syndrome) or without anosmia (n = 5) and SHFM, seven of whom (88%) harbor FGFR1 mutations. Of these seven, one individual is homozygous for p.V429E and six individuals are heterozygous for p.G348R, p.G485R, p.Q594*, p.E670A, p.V688L, or p.L712P. All mutations were predicted by in silico analysis to cause loss of function. Probands with FGFR1 mutations have severe gonadotropin-releasing hormone deficiency (absent puberty and/or cryptorchidism and/or micropenis). SHFM in both hands and feet was observed only in the patient with the homozygous p.V429E mutation; V429 maps to the fibroblast growth factor receptor substrate 2α binding domain of FGFR1, and functional studies of the p.V429E mutation demonstrated that it decreased recruitment and phosphorylation of fibroblast growth factor receptor substrate 2α to FGFR1, thereby resulting in reduced mitogen-activated protein kinase signaling.
Conclusion: FGFR1 should be prioritized for genetic testing in patients with CHH and SHFM because the likelihood of a mutation increases from 10% in the general CHH population to 88% in these patients.
Figures
Similar articles
-
FGFR1 Analyses in Four Patients with Hypogonadotropic Hypogonadism with Split-Hand/Foot Malformation: Implications for the Promoter Region.Hum Mutat. 2017 May;38(5):503-506. doi: 10.1002/humu.23178. Epub 2017 Feb 3. Hum Mutat. 2017. PMID: 28087897
-
A Novel FGFR1 Missense Mutation in a Portuguese Family with Congenital Hypogonadotropic Hypogonadism.Int J Mol Sci. 2022 Apr 17;23(8):4423. doi: 10.3390/ijms23084423. Int J Mol Sci. 2022. PMID: 35457241 Free PMC article.
-
Expanding the mutational spectrum of monogenic hypogonadotropic hypogonadism: novel mutations in ANOS1 and FGFR1 genes.Reprod Biol Endocrinol. 2020 Jan 29;18(1):8. doi: 10.1186/s12958-020-0568-6. Reprod Biol Endocrinol. 2020. PMID: 31996231 Free PMC article.
-
Congenital hypogonadotropic hypogonadism in females: clinical spectrum, evaluation and genetics.Ann Endocrinol (Paris). 2010 May;71(3):158-62. doi: 10.1016/j.ando.2010.02.024. Epub 2010 Apr 3. Ann Endocrinol (Paris). 2010. PMID: 20363464 Review.
-
Genetic regulatory pathways of split-hand/foot malformation.Clin Genet. 2019 Jan;95(1):132-139. doi: 10.1111/cge.13434. Epub 2018 Sep 10. Clin Genet. 2019. PMID: 30101460 Review.
Cited by
-
A novel dominant-negative FGFR1 variant causes Hartsfield syndrome by deregulating RAS/ERK1/2 pathway.Eur J Hum Genet. 2019 Jul;27(7):1113-1120. doi: 10.1038/s41431-019-0350-4. Epub 2019 Feb 20. Eur J Hum Genet. 2019. PMID: 30787447 Free PMC article.
-
Delayed Puberty-Phenotypic Diversity, Molecular Genetic Mechanisms, and Recent Discoveries.Endocr Rev. 2019 Oct 1;40(5):1285-1317. doi: 10.1210/er.2018-00248. Endocr Rev. 2019. PMID: 31220230 Free PMC article. Review.
-
The Differential Roles for Neurodevelopmental and Neuroendocrine Genes in Shaping GnRH Neuron Physiology and Deficiency.Int J Mol Sci. 2021 Aug 30;22(17):9425. doi: 10.3390/ijms22179425. Int J Mol Sci. 2021. PMID: 34502334 Free PMC article. Review.
-
Genetic testing facilitates prepubertal diagnosis of congenital hypogonadotropic hypogonadism.Clin Genet. 2017 Aug;92(2):213-216. doi: 10.1111/cge.12996. Epub 2017 Mar 30. Clin Genet. 2017. PMID: 28195315 Free PMC article.
-
[Molecular genetics and phenotypic features of congenital isolated hypogonadotropic hypogonadism].Probl Endokrinol (Mosk). 2021 Aug 6;67(4):46-56. doi: 10.14341/probl12787. Probl Endokrinol (Mosk). 2021. PMID: 34533013 Free PMC article. Russian.
References
-
- Tsai PS, Gill JC. Mechanisms of disease: Insights into X-linked and autosomal-dominant Kallmann syndrome. Nature clinical practice. Endocrinology & metabolism. 2006 Mar;2(3):160–171. - PubMed
-
- Dode C, Levilliers J, Dupont JM, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nature genetics. 2003 Apr;33(4):463–465. - PubMed
-
- Pitteloud N, Acierno JS, Jr., Meysing A, et al. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proceedings of the National Academy of Sciences of the United States of America. 2006 Apr 18;103(16):6281–6286. - PMC - PubMed
-
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine & growth factor reviews. 2005 Apr;16(2):139–149. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
