

Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency
- PMID: 24161539
- PMCID: PMC3898479
- DOI: 10.1016/j.bbadis.2013.10.008
Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency
Abstract
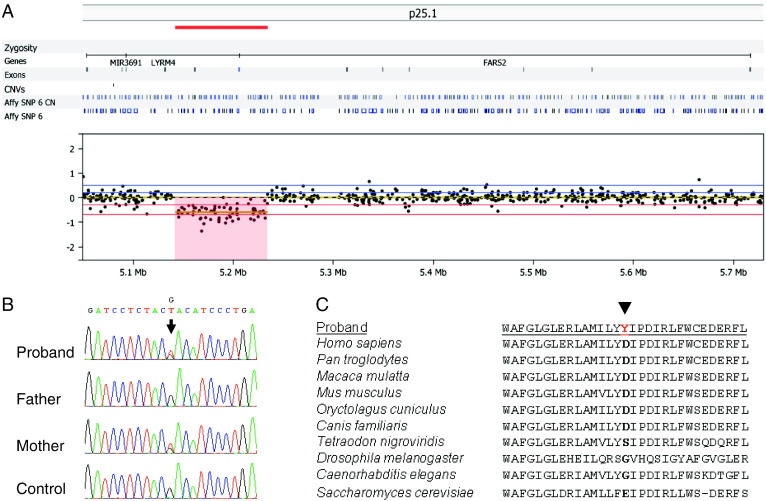
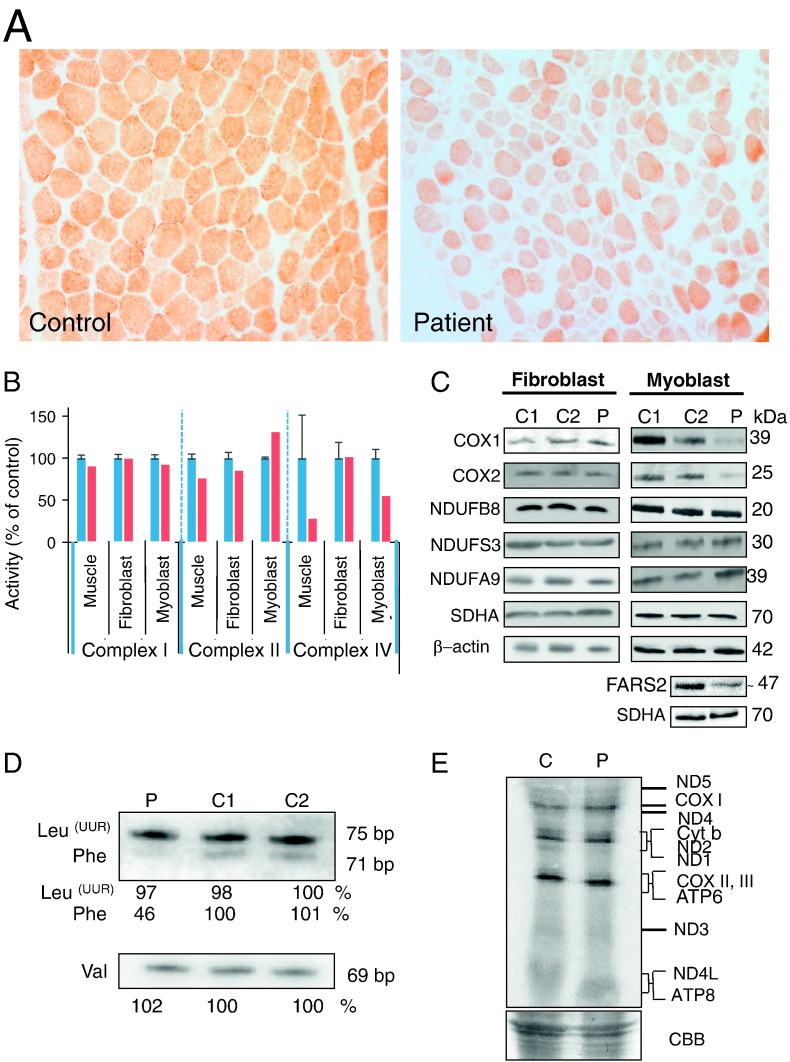
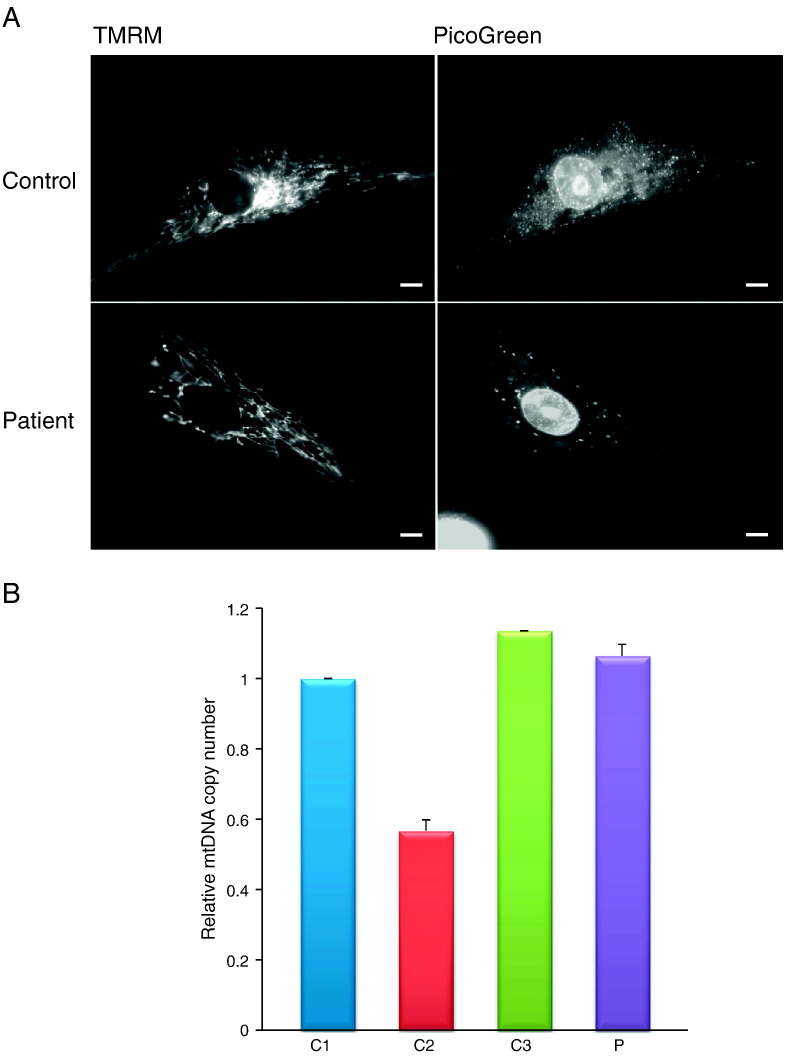
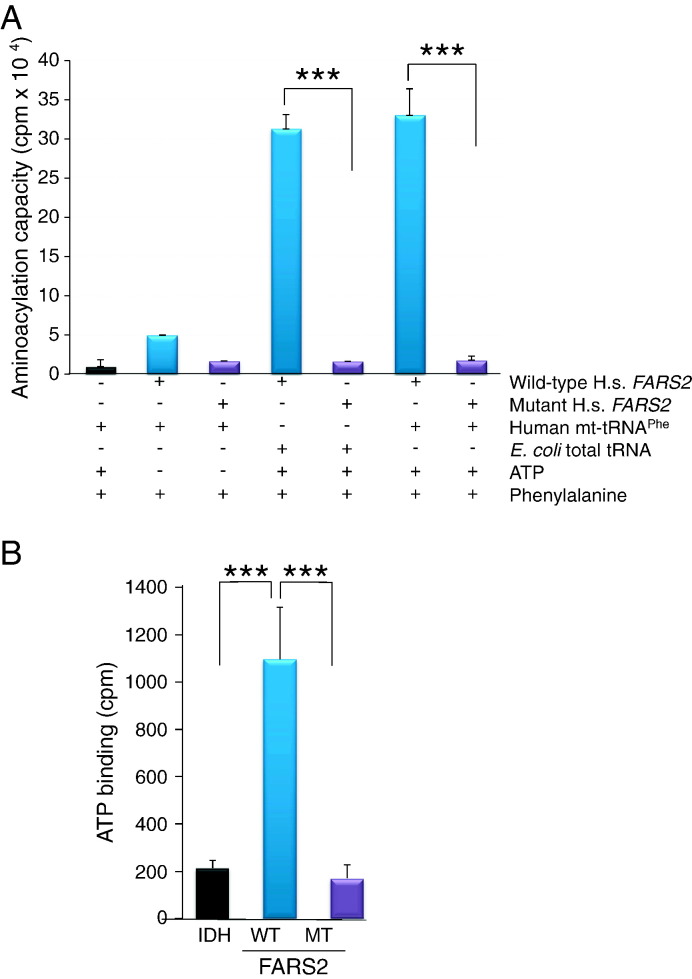
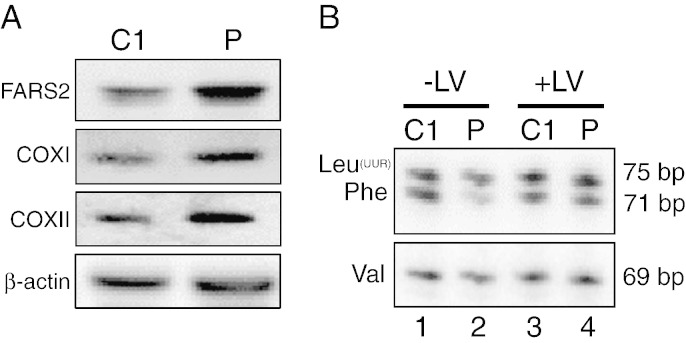
Mitochondrial aminoacyl-tRNA synthetases (aaRSs) are essential enzymes in protein synthesis since they charge tRNAs with their cognate amino acids. Mutations in the genes encoding mitochondrial aaRSs have been associated with a wide spectrum of human mitochondrial diseases. Here we report the identification of pathogenic mutations (a partial genomic deletion and a highly conserved p. Asp325Tyr missense variant) in FARS2, the gene encoding mitochondrial phenylalanyl-tRNA synthetase, in a patient with early-onset epilepsy and isolated complex IV deficiency in muscle. The biochemical defect was expressed in myoblasts but not in fibroblasts and associated with decreased steady state levels of COXI and COXII protein and reduced steady state levels of the mt-tRNA(Phe) transcript. Functional analysis of the recombinant mutant p. Asp325Tyr FARS2 protein showed an inability to bind ATP and consequently undetectable aminoacylation activity using either bacterial tRNA or human mt-tRNA(Phe) as substrates. Lentiviral transduction of cells with wildtype FARS2 restored complex IV protein levels, confirming that the p.Asp325Tyr mutation is pathogenic, causing respiratory chain deficiency and neurological deficits on account of defective aminoacylation of mt-tRNA(Phe).
Keywords: Aminoacyl-tRNA synthetase; Aminoacylation; LBSL; MLASA; MRI; Mitochondria; Mitochondrial disease; Mitochondrial translation; OXPHOS; PCH6; Protein synthesis; aaRS; aminoacyl-tRNA synthetase; leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation; magnetic resonance imaging; mitochondrial; mitochondrial DNA; mt-; mtDNA; myopathy, lactic acidosis and sideroblastic anaemia; oxidative phosphorylation; pontocerebellar hypoplasia type 6.
© 2013. Published by Elsevier B.V. All rights reserved.
Figures
Similar articles
-
Overexpression of mitochondrial histidyl-tRNA synthetase restores mitochondrial dysfunction caused by a deafness-associated tRNAHis mutation.J Biol Chem. 2020 Jan 24;295(4):940-954. doi: 10.1074/jbc.RA119.010998. Epub 2019 Dec 9. J Biol Chem. 2020. PMID: 31819004 Free PMC article.
-
New insights into the phenotype of FARS2 deficiency.Mol Genet Metab. 2017 Dec;122(4):172-181. doi: 10.1016/j.ymgme.2017.10.004. Epub 2017 Oct 12. Mol Genet Metab. 2017. PMID: 29126765 Free PMC article.
-
Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy.Hum Mol Genet. 2012 Oct 15;21(20):4521-9. doi: 10.1093/hmg/dds294. Epub 2012 Jul 23. Hum Mol Genet. 2012. PMID: 22833457
-
Role of Mutations of Mitochondrial Aminoacyl-tRNA Synthetases Genes on Epileptogenesis.Mol Neurobiol. 2023 Sep;60(9):5482-5492. doi: 10.1007/s12035-023-03429-1. Epub 2023 Jun 14. Mol Neurobiol. 2023. PMID: 37316759 Review.
-
Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases.Annu Rev Genet. 2011;45:299-329. doi: 10.1146/annurev-genet-110410-132531. Epub 2011 Sep 6. Annu Rev Genet. 2011. PMID: 21910628 Review.
Cited by
-
Neuropathy-associated Fars2 deficiency affects neuronal development and potentiates neuronal apoptosis by impairing mitochondrial function.Cell Biosci. 2022 Jul 6;12(1):103. doi: 10.1186/s13578-022-00838-y. Cell Biosci. 2022. PMID: 35794642 Free PMC article.
-
Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect.Am J Hum Genet. 2015 Apr 2;96(4):675-81. doi: 10.1016/j.ajhg.2015.02.012. Epub 2015 Mar 26. Am J Hum Genet. 2015. PMID: 25817015 Free PMC article.
-
Differential regulation of gene expression pathways with dexamethasone and ACTH after early life seizures.Neurobiol Dis. 2022 Nov;174:105873. doi: 10.1016/j.nbd.2022.105873. Epub 2022 Sep 21. Neurobiol Dis. 2022. PMID: 36152945 Free PMC article.
-
FARS2 mutations presenting with pure spastic paraplegia and lesions of the dentate nuclei.Ann Clin Transl Neurol. 2018 Aug 14;5(9):1128-1133. doi: 10.1002/acn3.598. eCollection 2018 Sep. Ann Clin Transl Neurol. 2018. PMID: 30250868 Free PMC article.
-
Expanding the clinical phenotype of IARS2-related mitochondrial disease.BMC Med Genet. 2018 Nov 12;19(1):196. doi: 10.1186/s12881-018-0709-3. BMC Med Genet. 2018. PMID: 30419932 Free PMC article.
References
-
- Tuppen H.A., Blakely E.L., Turnbull D.M., Taylor R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta. 2010;1797(2):113–128. - PubMed
-
- Rötig A. Human diseases with impaired mitochondrial protein synthesis. Biochim. Biophys. Acta Bioenerg. 2011;1807(9):1198–1205. - PubMed
-
- Bonnefond L., Fender A., Rudinger-Thirion J., Giege R., Florentz C., Sissler M. Toward the full set of human mitochondrial aminoacyl-tRNA synthetases: characterization of AspRS and TyrRS. Biochemistry. 2005;44(12):4805–4816. - PubMed
-
- Yadavalli S.S., Klipcan L., Zozulya A., Banerjee R., Svergun D., Safro M., Ibba M. Large-scale movement of functional domains facilitates aminoacylation by human mitochondrial phenylalanyl-tRNA synthetase. FEBS Lett. 2009;583(19):3204–3208. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
