

The phenotype and genotype of fibrodysplasia ossificans progressiva in China: a report of 72 cases
- PMID: 24051199
- PMCID: PMC3975922
- DOI: 10.1016/j.bone.2013.09.002
The phenotype and genotype of fibrodysplasia ossificans progressiva in China: a report of 72 cases
Abstract
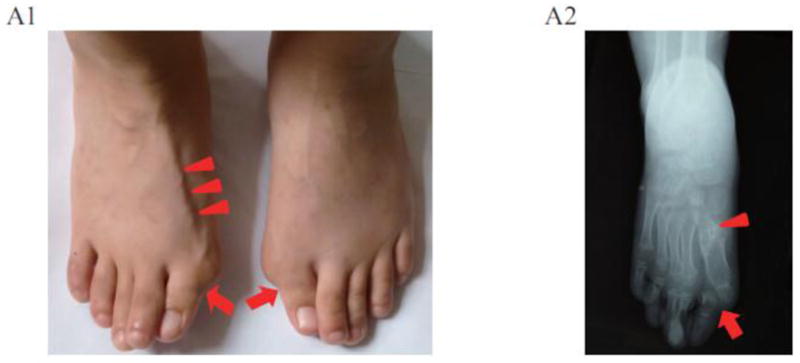
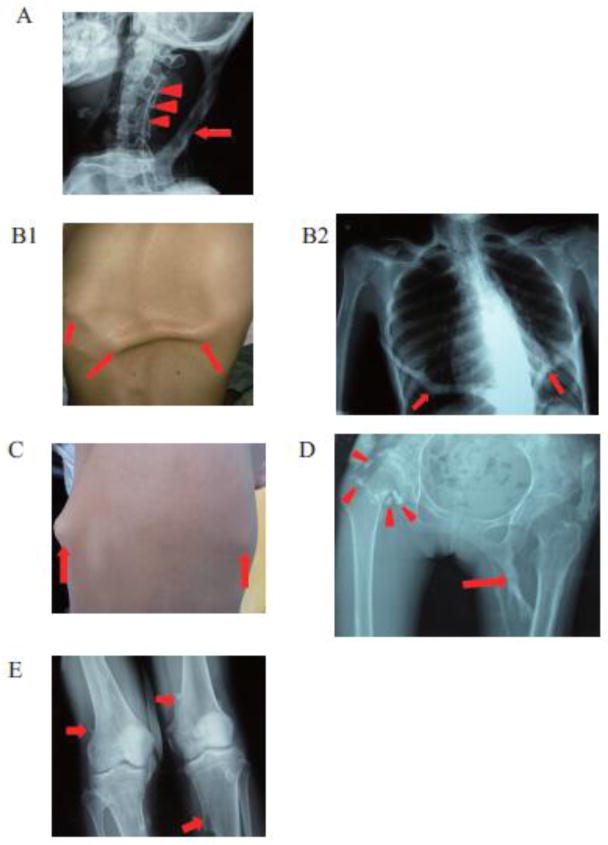
Fibrodysplasia ossificans progressiva, an ultra-rare and disabling genetic disorder of skeletal malformations and progressive heterotopic ossification (HO), is the most catastrophic condition of skeletal metamorphosis in humans. We studied 72 patients with FOP in China and analyzed their phenotypes and genotypes comprising the world's largest ethnically homogeneous population of FOP patients. Ninety-nine percent of patients (71/72 cases) were of Han nationality; and 1% of patients (1/72 cases) were of Hui nationality. Based on clinical examination, 92% of patients (66/72 cases) had classic FOP; 4% of patients (3/72 cases) were FOP-plus; and 4% of patients (3/72) were FOP variants. Importantly, all individuals with FOP had mutations in the protein-coding region of activin A receptor, type I/activin-like kinase 2 (ACVR1/ALK2). Ninety-seven percent of FOP patients (70/72 cases) had the canonical c.617G>A (p.R206H) mutation, while 3% of FOP patients (2/72 cases) had variant mutations in ACVR1/ALK2. Taken together, the genotypes and phenotypes of individuals with FOP from the Han nationality in China are similar to those reported elsewhere and support the fidelity of this ultra-rare disorder in the world's most highly populated nation and across wide racial, ethnic, gender and geographic distributions.
Keywords: ACVR1; ALK2; Bone morphogenetic protein; Fibrodysplasia ossificans progressiva; Heterotopic ossification.
© 2013.
Conflict of interest statement
None.
Figures
Similar articles
-
Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1.Hum Mutat. 2009 Mar;30(3):379-90. doi: 10.1002/humu.20868. Hum Mutat. 2009. PMID: 19085907 Free PMC article.
-
Depletion of Mast Cells and Macrophages Impairs Heterotopic Ossification in an Acvr1R206H Mouse Model of Fibrodysplasia Ossificans Progressiva.J Bone Miner Res. 2018 Feb;33(2):269-282. doi: 10.1002/jbmr.3304. Epub 2018 Jan 3. J Bone Miner Res. 2018. PMID: 28986986 Free PMC article.
-
Fibrodysplasia ossificans progressiva: clinical and genetic aspects.Orphanet J Rare Dis. 2011 Dec 1;6:80. doi: 10.1186/1750-1172-6-80. Orphanet J Rare Dis. 2011. PMID: 22133093 Free PMC article. Review.
-
Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice With the Human ACVR1(R206H) Fibrodysplasia Ossificans Progressiva (FOP) Mutation.J Bone Miner Res. 2016 Sep;31(9):1666-75. doi: 10.1002/jbmr.2820. Epub 2016 Mar 12. J Bone Miner Res. 2016. PMID: 26896819 Free PMC article.
-
Fibrodysplasia ossificans progressiva.Best Pract Res Clin Rheumatol. 2008 Mar;22(1):191-205. doi: 10.1016/j.berh.2007.11.007. Best Pract Res Clin Rheumatol. 2008. PMID: 18328989 Free PMC article. Review.
Cited by
-
Enhancer hijacking at the ARHGAP36 locus is associated with connective tissue to bone transformation.Nat Commun. 2023 Apr 11;14(1):2034. doi: 10.1038/s41467-023-37585-8. Nat Commun. 2023. PMID: 37041138 Free PMC article.
-
Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification.Sci Transl Med. 2016 Nov 23;8(366):366ra163. doi: 10.1126/scitranslmed.aaf1090. Sci Transl Med. 2016. PMID: 27881824 Free PMC article.
-
The biological function of type I receptors of bone morphogenetic protein in bone.Bone Res. 2016 Apr 5;4:16005. doi: 10.1038/boneres.2016.5. eCollection 2016. Bone Res. 2016. PMID: 27088043 Free PMC article. Review.
-
Fibrodysplasia ossificans progressiva: Basic understanding and experimental models.Intractable Rare Dis Res. 2017 Nov;6(4):242-248. doi: 10.5582/irdr.2017.01055. Intractable Rare Dis Res. 2017. PMID: 29259851 Free PMC article. Review.
-
Analysis of clinical manifestations and treatment in 26 children with fibrodysplasia ossificans progressiva in China.World J Pediatr. 2020 Feb;16(1):82-88. doi: 10.1007/s12519-019-00302-x. Epub 2019 Sep 16. World J Pediatr. 2020. PMID: 31529313
References
-
- Shore EM, Feldman GJ, Xu M, Kaplan FS. The genetics of fibrodysplasia ossificans progressiva. Clin Rev Bone & Miner Metab. 2005;3:201–4.
-
- Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, et al. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressive. J Bone Joint Surg Am. 1993;75:215–9. - PubMed
-
- Kitterman JA, Kantanie S, Rocke DM, Kaplan FS. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics. 2005;116:654–61. - PubMed
-
- Kaplan FS, Glaser DL. Thoracic insufficiency syndrome in patients with fibrodysplasia ossificans progressiva. Clin Rev Bone & Miner Metab. 2005;3:213–6.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
