

Alternative splicing modifies the effect of mutations in COL11A1 and results in recessive type 2 Stickler syndrome with profound hearing loss
- PMID: 23922384
- PMCID: PMC3812854
- DOI: 10.1136/jmedgenet-2012-101499
Alternative splicing modifies the effect of mutations in COL11A1 and results in recessive type 2 Stickler syndrome with profound hearing loss
Abstract
Background: Stickler syndromes types 1, 2 and 3 are usually dominant disorders caused by mutations in the genes COL2A1, COL11A1 and COL11A2 that encode the fibrillar collagens types II and XI present in cartilage and vitreous. Rare recessive forms of Stickler syndrome exist that are due to mutations in genes encoding type IX collagen (COL9A1 type 4 Stickler syndrome and COL9A2 type 5 Stickler syndrome). Recently, recessive mutations in the COL11A1 gene have been demonstrated to result in fibrochondrogenesis, a much more severe skeletal dysplasia, which is often lethal. Here we demonstrate that some mutations in COL11A1 are recessive, modified by alternative splicing and result in type 2 Stickler syndrome rather than fibrochondrogenesis.
Methods: Patients referred to the national Stickler syndrome diagnostic service for England, UK were assessed clinically and subsequently sequenced for mutations in COL11A1. Additional in silico and functional studies to assess the effect of sequence variants on pre-mRNA processing and collagen structure were performed.
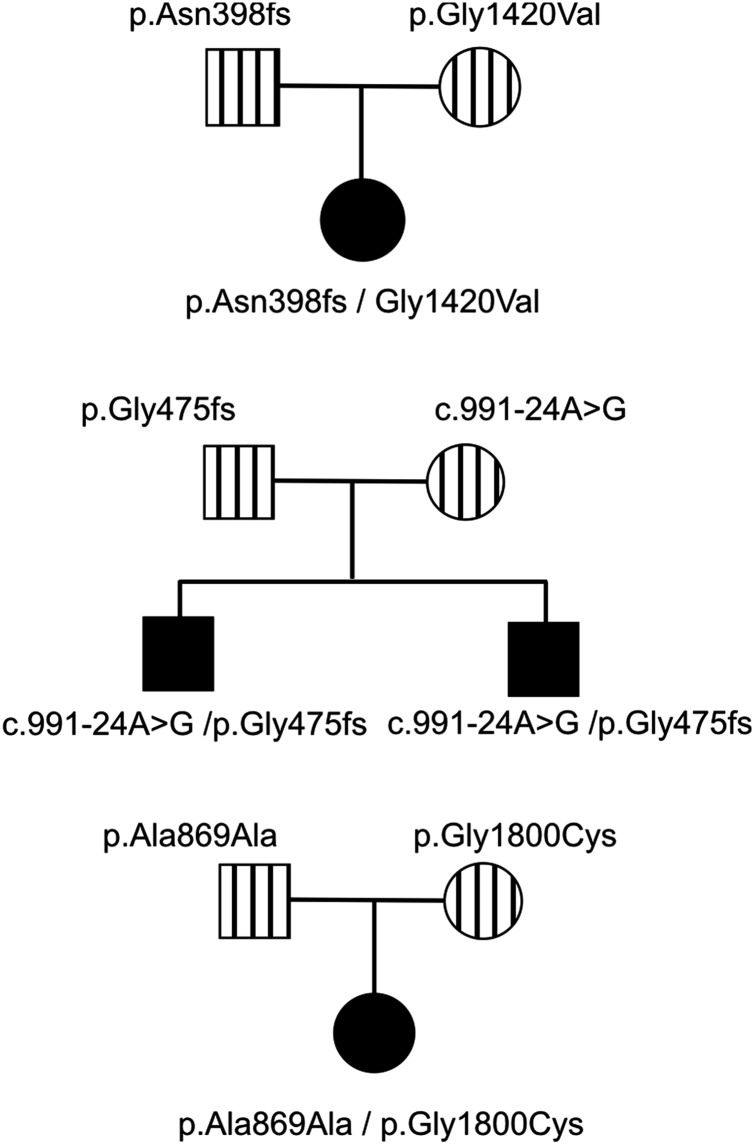
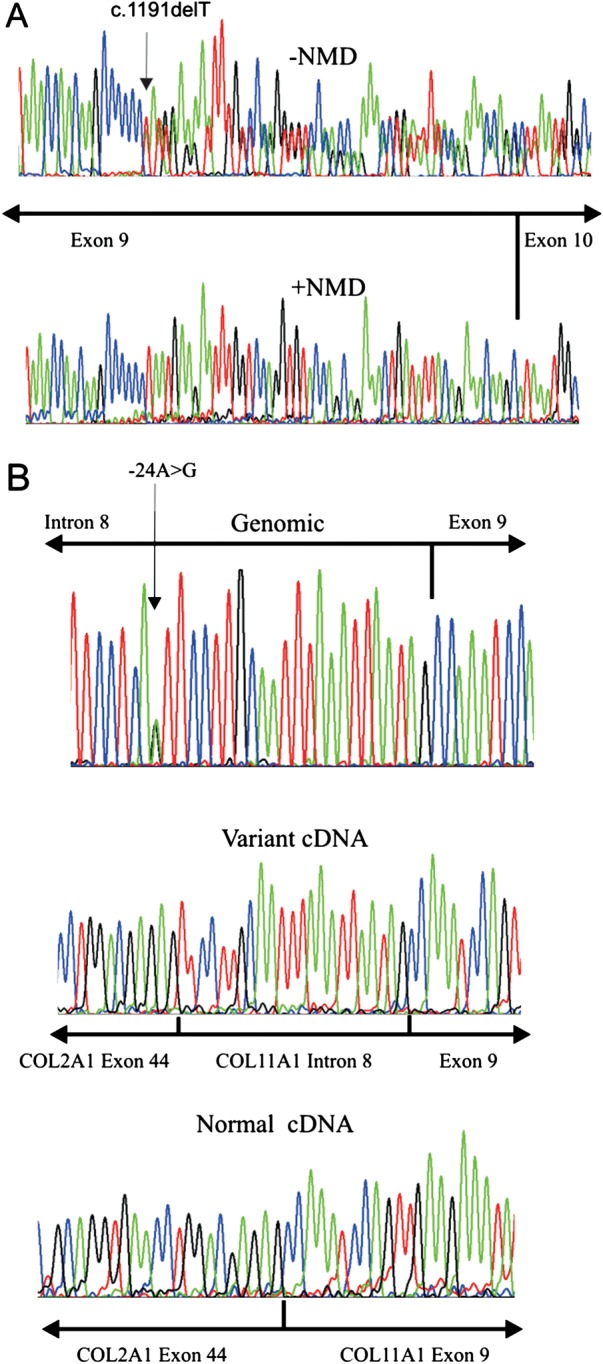
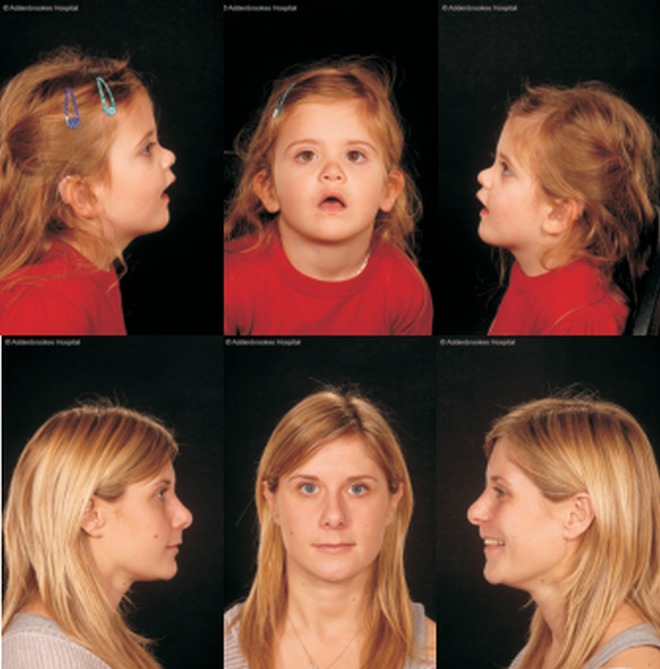
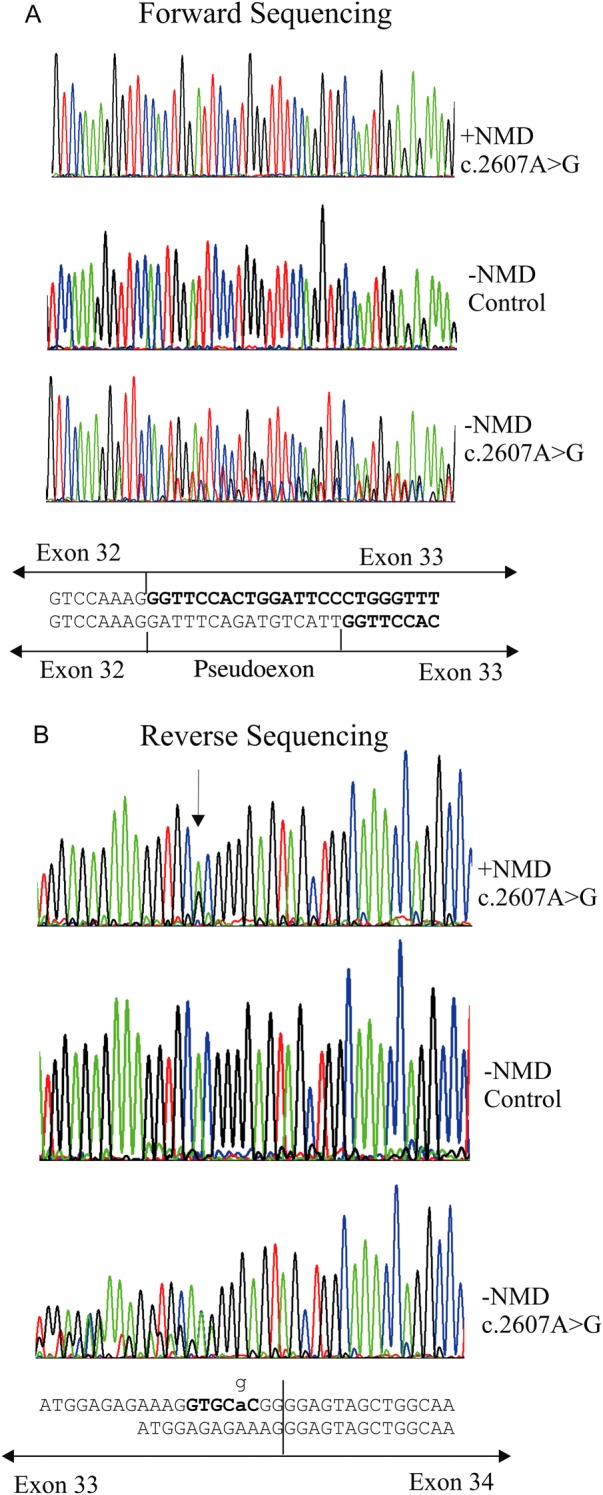
Results: In three different families, heterozygous COL11A1 biallelic null, null/missense or silent/missense mutations, were found. They resulted in a recessive form of type 2 Stickler syndrome characterised by particularly profound hearing loss and are clinically distinct from the recessive types 4 and 5 variants of Stickler syndrome. One mutant allele in each family is capable of synthesising a normal α1(XI) procollagen molecule, via variable pre-mRNA processing.
Conclusion: This new variant has important implications for molecular diagnosis and counselling families with type 2 Stickler syndrome.
Keywords: Alternative Splicing; Recessive Inheritance; Stickler syndrome.
Figures

Similar articles
-
Inherited and de novo biallelic pathogenic variants in COL11A1 result in type 2 Stickler syndrome with severe hearing loss.Mol Genet Genomic Med. 2020 Sep;8(9):e1354. doi: 10.1002/mgg3.1354. Epub 2020 Jun 24. Mol Genet Genomic Med. 2020. PMID: 32578940 Free PMC article.
-
Exon-Trapping Assay Improves Clinical Interpretation of COL11A1 and COL11A2 Intronic Variants in Stickler Syndrome Type 2 and Otospondylomegaepiphyseal Dysplasia.Genes (Basel). 2020 Dec 17;11(12):1513. doi: 10.3390/genes11121513. Genes (Basel). 2020. PMID: 33348901 Free PMC article.
-
A novel dominant COL11A1 mutation in a child with Stickler syndrome type II is associated with recurrent fractures.Osteoporos Int. 2018 Jan;29(1):247-251. doi: 10.1007/s00198-017-4229-3. Epub 2017 Oct 3. Osteoporos Int. 2018. PMID: 28971234
-
Hearing impairment in Stickler syndrome: a systematic review.Orphanet J Rare Dis. 2012 Oct 30;7:84. doi: 10.1186/1750-1172-7-84. Orphanet J Rare Dis. 2012. PMID: 23110709 Free PMC article. Review.
-
A novel mutation in the COL2A1 gene in a patient with Stickler syndrome type 1: a case report and review of the literature.J Med Case Rep. 2017 Aug 26;11(1):237. doi: 10.1186/s13256-017-1396-y. J Med Case Rep. 2017. PMID: 28841907 Free PMC article. Review.
Cited by
-
Dominant Stickler Syndrome.Genes (Basel). 2022 Jun 18;13(6):1089. doi: 10.3390/genes13061089. Genes (Basel). 2022. PMID: 35741851 Free PMC article. Review.
-
Inherited and de novo biallelic pathogenic variants in COL11A1 result in type 2 Stickler syndrome with severe hearing loss.Mol Genet Genomic Med. 2020 Sep;8(9):e1354. doi: 10.1002/mgg3.1354. Epub 2020 Jun 24. Mol Genet Genomic Med. 2020. PMID: 32578940 Free PMC article.
-
COL11A1 is associated with developmental dysplasia of the hip and secondary osteoarthritis in the HUNT study.Osteoarthr Cartil Open. 2023 Dec 16;6(1):100424. doi: 10.1016/j.ocarto.2023.100424. eCollection 2024 Mar. Osteoarthr Cartil Open. 2023. PMID: 38283578 Free PMC article.
-
Genetic architecture distinguishes tinnitus from hearing loss.Nat Commun. 2024 Jan 19;15(1):614. doi: 10.1038/s41467-024-44842-x. Nat Commun. 2024. PMID: 38242899 Free PMC article.
-
Autosomal recessive Stickler syndrome resulting from a COL9A3 mutation.Am J Med Genet A. 2018 Dec;176(12):2887-2891. doi: 10.1002/ajmg.a.40647. Epub 2018 Nov 18. Am J Med Genet A. 2018. PMID: 30450842 Free PMC article. Review.
References
-
- Burgeson RE, Hollister DW. Collagen heterogeneity in human cartilage: identification of several new collagen chains. Biochem Biophys Res Commun 1979;87:1124–31 - PubMed
-
- Morris NP, Bächinger HP. Type XI collagen is a heterotrimer with the composition (1α,2α,3α) retaining non-triple-helical domains. J Biol Chem 1987;262:11345–50 - PubMed
-
- Bernard M, Yoshioka H, Rodriguez E, Van der Rest M, Kimura T, Ninomiya Y, Olsen BR, Ramirez F. Cloning and sequencing of pro-α1 (XI) collagen cDNA demonstrates that type XI belongs to the fibrillar class of collagens and reveals that the expression of the gene is not restricted to cartilagenous tissue. J Biol Chem 1988;263:17159–66 - PubMed
-
- Kimura T, Cheah KS, Chan SD, Lui VC, Mattei MG, van der Rest M, Ono K, Solomon E, Ninomiya Y, Olsen BR. The human alpha 2(XI) collagen (COL11A2) chain. Molecular cloning of cDNA and genomic DNA reveals characteristics of a fibrillar collagen with differences in genomic organization. J Biol Chem 1989;264:13910–16 - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous