

Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly
- PMID: 23603762
- PMCID: PMC3826256
- DOI: 10.1038/ng.2613
Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly
Erratum in
- Nat Genet. 2013 Aug;45(8):962
Abstract
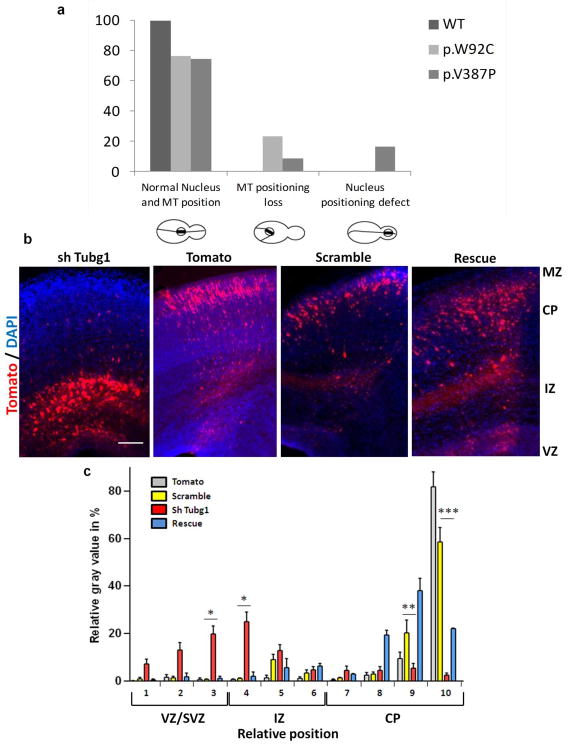
The genetic causes of malformations of cortical development (MCD) remain largely unknown. Here we report the discovery of multiple pathogenic missense mutations in TUBG1, DYNC1H1 and KIF2A, as well as a single germline mosaic mutation in KIF5C, in subjects with MCD. We found a frequent recurrence of mutations in DYNC1H1, implying that this gene is a major locus for unexplained MCD. We further show that the mutations in KIF5C, KIF2A and DYNC1H1 affect ATP hydrolysis, productive protein folding and microtubule binding, respectively. In addition, we show that suppression of mouse Tubg1 expression in vivo interferes with proper neuronal migration, whereas expression of altered γ-tubulin proteins in Saccharomyces cerevisiae disrupts normal microtubule behavior. Our data reinforce the importance of centrosomal and microtubule-related proteins in cortical development and strongly suggest that microtubule-dependent mitotic and postmitotic processes are major contributors to the pathogenesis of MCD.
Figures
Similar articles
-
Beta tubulin isoforms are not interchangeable for rescuing impaired radial migration due to Tubb3 knockdown.Hum Mol Genet. 2014 Mar 15;23(6):1516-26. doi: 10.1093/hmg/ddt538. Epub 2013 Oct 30. Hum Mol Genet. 2014. PMID: 24179174
-
Exome Sequencing Identifies De Novo DYNC1H1 Mutations Associated With Distal Spinal Muscular Atrophy and Malformations of Cortical Development.J Child Neurol. 2017 Mar;32(4):379-386. doi: 10.1177/0883073816683083. Epub 2016 Dec 20. J Child Neurol. 2017. PMID: 28193117
-
TUBG1 missense variants underlying cortical malformations disrupt neuronal locomotion and microtubule dynamics but not neurogenesis.Nat Commun. 2019 May 13;10(1):2129. doi: 10.1038/s41467-019-10081-8. Nat Commun. 2019. PMID: 31086189 Free PMC article.
-
Tubulin mutations in human neurodevelopmental disorders.Semin Cell Dev Biol. 2023 Mar 15;137:87-95. doi: 10.1016/j.semcdb.2022.07.009. Epub 2022 Jul 30. Semin Cell Dev Biol. 2023. PMID: 35915025 Review.
-
Tubulin genes and malformations of cortical development.Eur J Med Genet. 2018 Dec;61(12):744-754. doi: 10.1016/j.ejmg.2018.07.012. Epub 2018 Jul 17. Eur J Med Genet. 2018. PMID: 30016746 Review.
Cited by
-
The clinical-phenotype continuum in DYNC1H1-related disorders-genomic profiling and proposal for a novel classification.J Hum Genet. 2020 Nov;65(11):1003-1017. doi: 10.1038/s10038-020-0803-1. Epub 2020 Aug 12. J Hum Genet. 2020. PMID: 32788638 Free PMC article.
-
Mechanisms and functions of lysosome positioning.J Cell Sci. 2016 Dec 1;129(23):4329-4339. doi: 10.1242/jcs.196287. Epub 2016 Oct 31. J Cell Sci. 2016. PMID: 27799357 Free PMC article. Review.
-
Non-centrosomal nucleation mediated by augmin organizes microtubules in post-mitotic neurons and controls axonal microtubule polarity.Nat Commun. 2016 Jul 13;7:12187. doi: 10.1038/ncomms12187. Nat Commun. 2016. PMID: 27405868 Free PMC article.
-
CAMSAPs organize an acentrosomal microtubule network from basal varicosities in radial glial cells.J Cell Biol. 2021 Aug 2;220(8):e202003151. doi: 10.1083/jcb.202003151. Epub 2021 May 21. J Cell Biol. 2021. PMID: 34019079 Free PMC article.
-
Aging Alters the Aortic Proteome in Health and Thoracic Aortic Aneurysm.Arterioscler Thromb Vasc Biol. 2022 Aug;42(8):1060-1076. doi: 10.1161/ATVBAHA.122.317643. Epub 2022 May 5. Arterioscler Thromb Vasc Biol. 2022. PMID: 35510553 Free PMC article.
References
-
- Caviness VS, Jr, Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–83. - PubMed
-
- Rakic P, Caviness VS., Jr Cortical development: view from neurological mutants two decades later. Neuron. 1995;14:1101–4. - PubMed
-
- Francis F, et al. Human disorders of cortical development: from past to present. Eur J Neurosci. 2006;23:877–93. - PubMed
-
- Guerrini R, Dobyns WB, Barkovich AJ. Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. Trends Neurosci. 2008;31:154–62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
