

Update on Kleefstra Syndrome
- PMID: 22670141
- PMCID: PMC3366700
- DOI: 10.1159/000335648
Update on Kleefstra Syndrome
Abstract
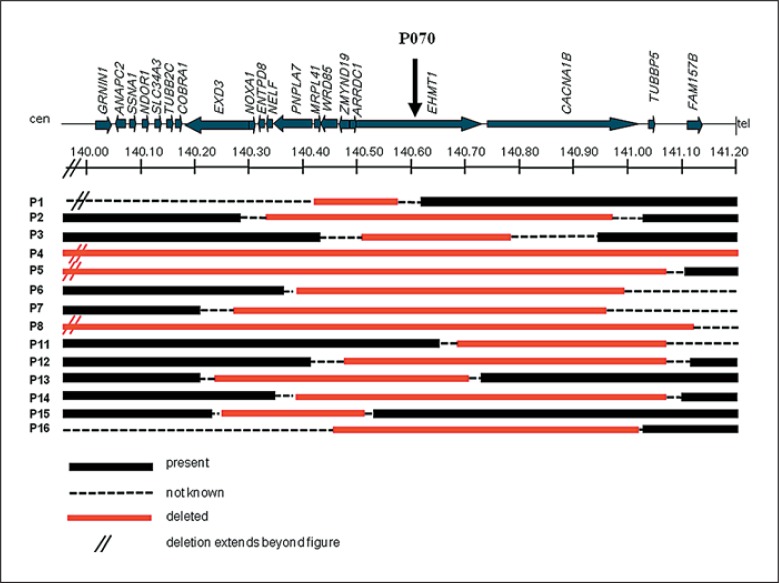
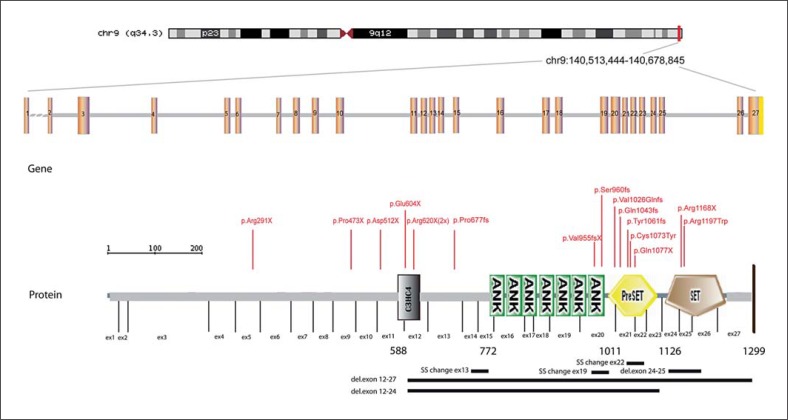
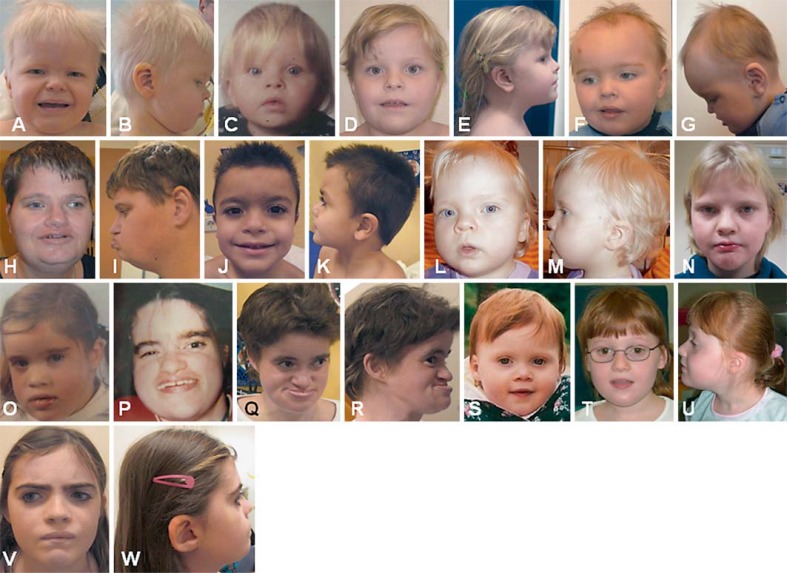
Kleefstra syndrome is characterized by the core phenotype of developmental delay/intellectual disability, (childhood) hypotonia and distinct facial features. The syndrome can be either caused by a microdeletion in chromosomal region 9q34.3 or by a mutation in the euchromatin histone methyltransferase 1 (EHMT1) gene. Since the early 1990s, 85 patients have been described, of which the majority had a 9q34.3 microdeletion (>85%). So far, no clear genotype-phenotype correlation could be observed by studying the clinical and molecular features of both 9q34.3 microdeletion patients and patients with an intragenic EHMT1 mutation. Thus, to further expand the genotypic and phenotypic knowledge about the syndrome, we here report 29 newly diagnosed patients, including 16 patients with a 9q34.3 microdeletion and 13 patients with an EHMT1 mutation, and review previous literature. The present findings are comparable to previous reports. In addition to our former findings and recommendations, we suggest cardiac screening during follow-up, because of the possible occurrence of cardiac arrhythmias. In addition, clinicians and caretakers should be aware of the regressive behavioral phenotype that might develop at adolescent/adult age and seems to have no clear neurological substrate, but is rather a so far unexplained neuropsychiatric feature.
Figures

Similar articles
-
Pulmonary hypertension in patients with 9q34.3 microdeletion-associated Kleefstra syndrome.Am J Med Genet A. 2018 Aug;176(8):1773-1777. doi: 10.1002/ajmg.a.38852. Epub 2018 Jul 31. Am J Med Genet A. 2018. PMID: 30063093 Free PMC article.
-
Intragenic duplication of EHMT1 gene results in Kleefstra syndrome.Mol Cytogenet. 2014 Oct 23;7(1):74. doi: 10.1186/s13039-014-0074-7. eCollection 2014. Mol Cytogenet. 2014. PMID: 25349628 Free PMC article.
-
Severe neonatal presentation of Kleefstra syndrome in a patient with hypoplastic left heart syndrome and 9q34.3 microdeletion.Birth Defects Res A Clin Mol Teratol. 2014 Dec;100(12):985-90. doi: 10.1002/bdra.23324. Epub 2014 Nov 7. Birth Defects Res A Clin Mol Teratol. 2014. PMID: 25380126
-
First prenatal diagnosis of a 'pure' 9q34.3 deletion (Kleefstra syndrome): A case report and literature review.J Obstet Gynaecol Res. 2018 Mar;44(3):570-575. doi: 10.1111/jog.13517. Epub 2017 Nov 21. J Obstet Gynaecol Res. 2018. PMID: 29160022 Review.
-
New Insights into Kleefstra Syndrome: Report of Two Novel Cases with Previously Unreported Features and Literature Review.Cytogenet Genome Res. 2018;156(3):127-133. doi: 10.1159/000494532. Epub 2018 Nov 17. Cytogenet Genome Res. 2018. PMID: 30448833 Review.
Cited by
-
CRISPR single base editing, neuronal disease modelling and functional genomics for genetic variant analysis: pipeline validation using Kleefstra syndrome EHMT1 haploinsufficiency.Stem Cell Res Ther. 2022 Feb 9;13(1):69. doi: 10.1186/s13287-022-02740-3. Stem Cell Res Ther. 2022. PMID: 35139903 Free PMC article.
-
Biochemical validation of EHMT1 missense mutations in Kleefstra syndrome.J Hum Genet. 2018 May;63(5):555-562. doi: 10.1038/s10038-018-0413-3. Epub 2018 Feb 19. J Hum Genet. 2018. PMID: 29459631
-
9q34.3 microduplications lead to neurodevelopmental disorders through EHMT1 overexpression.Neurogenetics. 2019 Aug;20(3):145-154. doi: 10.1007/s10048-019-00581-6. Epub 2019 Jun 17. Neurogenetics. 2019. PMID: 31209758
-
Genetic studies in intellectual disability and related disorders.Nat Rev Genet. 2016 Jan;17(1):9-18. doi: 10.1038/nrg3999. Epub 2015 Oct 27. Nat Rev Genet. 2016. PMID: 26503795 Review.
-
Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis.J Clin Invest. 2015 Feb;125(2):478-86. doi: 10.1172/JCI78362. Epub 2015 Feb 2. J Clin Invest. 2015. PMID: 25642708 Free PMC article. Review.
References
-
- Dawson AJ, Putnam S, Schultz J, Riordan D, Prasad C, et al. Cryptic chromosome rearrangements detected by subtelomere assay in patients with mental retardation and dysmorphic features. Clin Genet. 2002;62:488–494. - PubMed
-
- Font-Montgomery E, Weaver DD, Walsh L, Christensen C, Thurston VC. Clinical and cytogenetic manifestations of subtelomeric aberrations: report of six cases. Birth Defects Res A Clin Mol Teratol. 2004;70:408–415. - PubMed
-
- Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. - PubMed
LinkOut - more resources
Full Text Sources
