

Novel C16orf57 mutations in patients with Poikiloderma with Neutropenia: bioinformatic analysis of the protein and predicted effects of all reported mutations
- PMID: 22269211
- PMCID: PMC3315733
- DOI: 10.1186/1750-1172-7-7
Novel C16orf57 mutations in patients with Poikiloderma with Neutropenia: bioinformatic analysis of the protein and predicted effects of all reported mutations
Abstract
Background: Poikiloderma with Neutropenia (PN) is a rare autosomal recessive genodermatosis caused by C16orf57 mutations. To date 17 mutations have been identified in 31 PN patients.
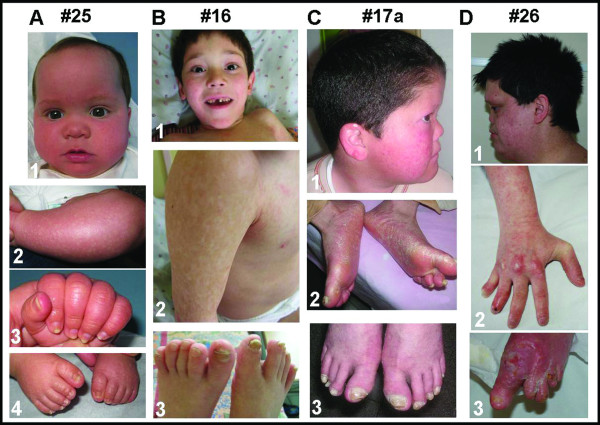
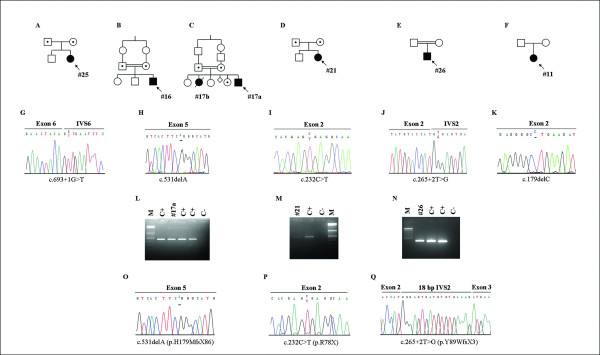
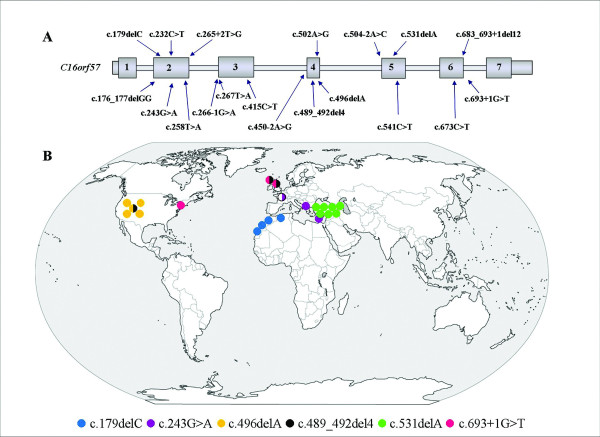
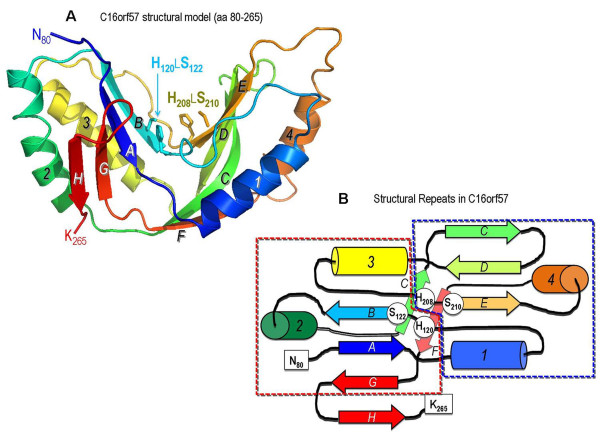
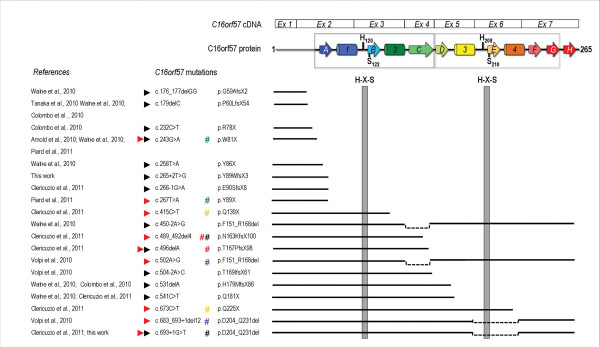
Results: We characterize six PN patients expanding the clinical phenotype of the syndrome and the mutational repertoire of the gene. We detect the two novel C16orf57 mutations, c.232C>T and c.265+2T>G, as well as the already reported c.179delC, c.531delA and c.693+1G>T mutations. cDNA analysis evidences the presence of aberrant transcripts, and bioinformatic prediction of C16orf57 protein structure gauges the mutations effects on the folded protein chain. Computational analysis of the C16orf57 protein shows two conserved H-X-S/T-X tetrapeptide motifs marking the active site of a two-fold pseudosymmetric structure recalling the 2H phosphoesterase superfamily. Based on this model C16orf57 is likely a 2H-active site enzyme functioning in RNA processing, as a presumptive RNA ligase. According to bioinformatic prediction, all known C16orf57 mutations, including the novel mutations herein described, impair the protein structure by either removing one or both tetrapeptide motifs or by destroying the symmetry of the native folding.Finally, we analyse the geographical distribution of the recurrent mutations that depicts clusters featuring a founder effect.
Conclusions: In cohorts of patients clinically affected by genodermatoses with overlapping symptoms, the molecular screening of C16orf57 gene seems the proper way to address the correct diagnosis of PN, enabling the syndrome-specific oncosurveillance. The bioinformatic prediction of the C16orf57 protein structure denotes a very basic enzymatic function consistent with a housekeeping function. Detection of aberrant transcripts, also in cells from PN patients carrying early truncated mutations, suggests they might be translatable. Tissue-specific sensitivity to the lack of functionally correct protein accounts for the main cutaneous and haematological clinical signs of PN patients.
Figures
Similar articles
-
Identification of a novel C16orf57 mutation in Athabaskan patients with Poikiloderma with Neutropenia.Am J Med Genet A. 2011 Feb;155A(2):337-42. doi: 10.1002/ajmg.a.33807. Epub 2010 Dec 22. Am J Med Genet A. 2011. PMID: 21271650 Free PMC article.
-
Poikiloderma with neutropenia: a novel C16orf57 mutation and clinical diagnostic criteria.Br J Dermatol. 2010 Oct;163(4):866-9. doi: 10.1111/j.1365-2133.2010.09929.x. Epub 2010 Sep 7. Br J Dermatol. 2010. PMID: 20618321
-
Identification of a homozygous deletion mutation in C16orf57 in a family with Clericuzio-type poikiloderma with neutropenia.Am J Med Genet A. 2010 Jun;152A(6):1347-8. doi: 10.1002/ajmg.a.33455. Am J Med Genet A. 2010. PMID: 20503306 No abstract available.
-
Poikiloderma with neutropenia: a case report and review of the literature.J Pediatr Hematol Oncol. 2014 May;36(4):297-300. doi: 10.1097/MPH.0b013e31829f35e7. J Pediatr Hematol Oncol. 2014. PMID: 23823120 Review.
-
Inherited skin disorders presenting with poikiloderma.Int J Dermatol. 2021 Nov;60(11):1343-1353. doi: 10.1111/ijd.15498. Epub 2021 Mar 19. Int J Dermatol. 2021. PMID: 33739439 Review.
Cited by
-
Insights into Mutation Effect in Three Poikiloderma with Neutropenia Patients by Transcript Analysis and Disease Evolution of Reported Patients with the Same Pathogenic Variants.J Clin Immunol. 2018 May;38(4):494-502. doi: 10.1007/s10875-018-0508-9. Epub 2018 May 16. J Clin Immunol. 2018. PMID: 29770900
-
Primary Immunodeficiencies With Defects in Innate Immunity: Focus on Orofacial Manifestations.Front Immunol. 2020 Jun 18;11:1065. doi: 10.3389/fimmu.2020.01065. eCollection 2020. Front Immunol. 2020. PMID: 32625202 Free PMC article. Review.
-
One Disease, Many Genes: Implications for the Treatment of Osteopetroses.Front Endocrinol (Lausanne). 2019 Feb 19;10:85. doi: 10.3389/fendo.2019.00085. eCollection 2019. Front Endocrinol (Lausanne). 2019. PMID: 30837952 Free PMC article. Review.
-
Poikiloderma With Neutropenia and Mastocytosis: A Case Report and a Review of Dermatological Signs.Front Med (Lausanne). 2021 Jun 10;8:680363. doi: 10.3389/fmed.2021.680363. eCollection 2021. Front Med (Lausanne). 2021. PMID: 34179048 Free PMC article.
-
Clinical utility gene card for: poikiloderma with neutropenia.Eur J Hum Genet. 2013 Oct;21(10). doi: 10.1038/ejhg.2012.298. Epub 2013 Jan 16. Eur J Hum Genet. 2013. PMID: 23321617 Free PMC article. No abstract available.
References
-
- Clericuzio C, Hoyme HE, Asse JM. Immune deficient poikiloderma: a new genodermatosis. AJHG. 1991;49(Suppl):A661.
-
- Concolino D, Roversi G, Muzzi GL, Sestito S, Colombo EA, Volpi L, Larizza L, Strisciuglio P. Clericuzio type Poikiloderma with Neutropenia Syndrome in three sibs with mutations in the C16orf57 gene: delineation of the phenotype. Am J Med Genet A. 2010;152A:2588–2594. doi: 10.1002/ajmg.a.33600. - DOI - PubMed
