

Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias
- PMID: 22120146
- PMCID: PMC3235563
- DOI: 10.1093/brain/awr289
Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias
Abstract
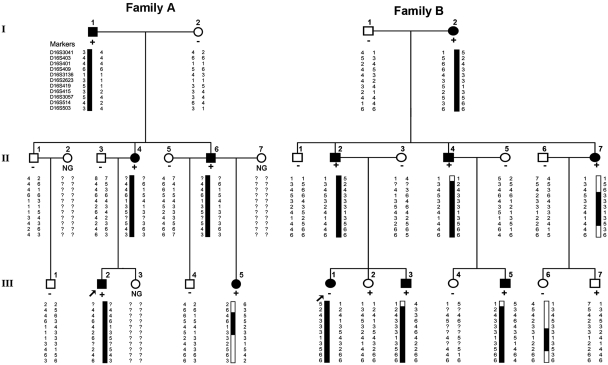
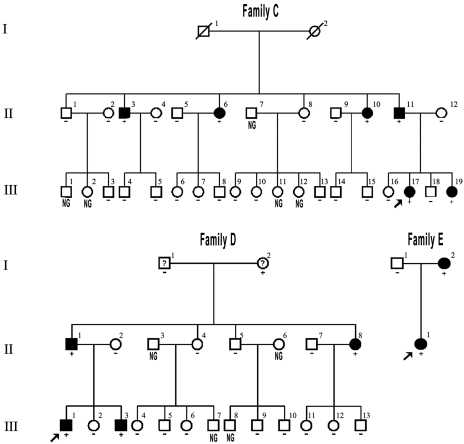
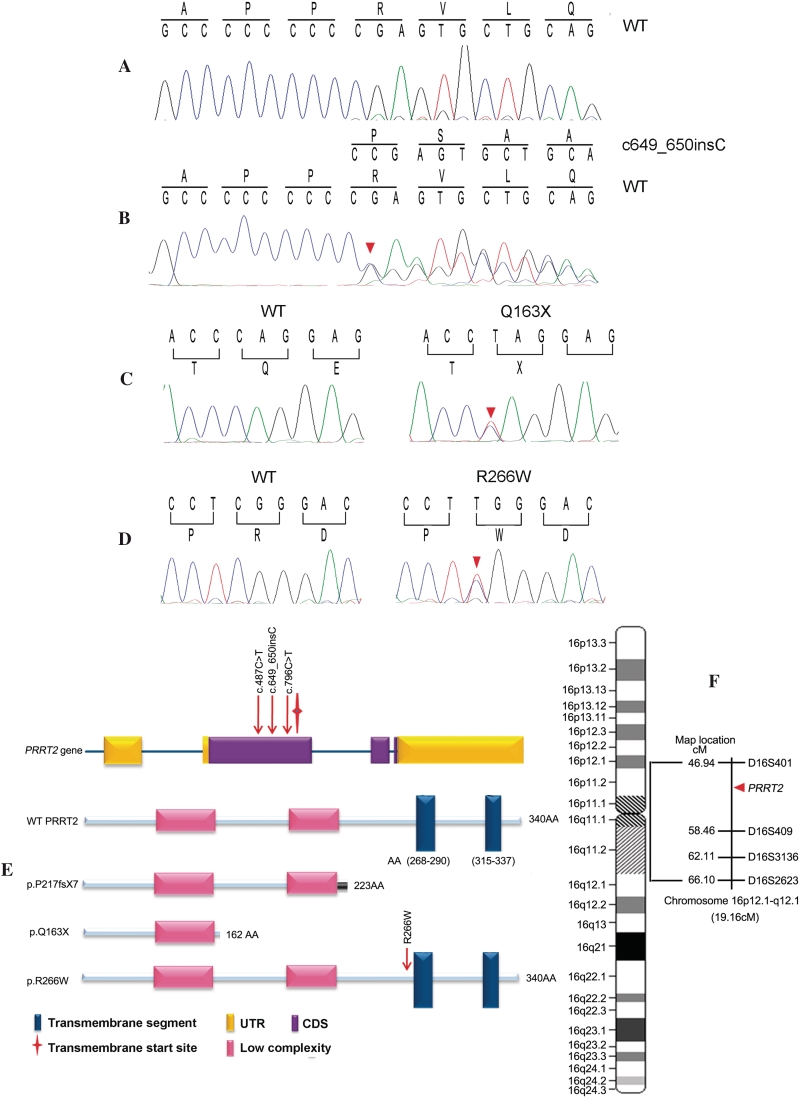
Paroxysmal kinesigenic dyskinesias is a paroxysmal movement disorder characterized by recurrent, brief attacks of abnormal involuntary movements induced by sudden voluntary movements. Although several loci, including the pericentromeric region of chromosome 16, have been linked to paroxysmal kinesigenic dyskinesias, the causative gene has not yet been identified. Here, we identified proline-rich transmembrane protein 2 (PRRT2) as a causative gene of paroxysmal kinesigenic dyskinesias by using a combination of exome sequencing and linkage analysis. Genetic linkage mapping with 11 markers that encompassed the pericentromeric of chromosome 16 was performed in 27 members of two families with autosomal dominant paroxysmal kinesigenic dyskinesias. Then, the whole-exome sequencing was performed in three patients from these two families. By combining the defined linkage region (16p12.1-q12.1) and the results of exome sequencing, we identified an insertion mutation c.649_650InsC (p.P217fsX7) in one family and a nonsense mutation c.487C>T (p.Q163X) in another family. To confirm our findings, we sequenced the exons and flanking introns of PRRT2 in another three families with paroxysmal kinesigenic dyskinesias. The c.649_650InsC (p.P217fsX7) mutation was identified in two of these families, whereas a missense mutation, c.796C>T (R266W), was identified in another family with paroxysmal kinesigenic dyskinesias. All of these mutations completely co-segregated with the phenotype in each family. None of these mutations was identified in 500 normal unaffected individuals of matched geographical ancestry. Thus, we have identified PRRT2 as the first causative gene of paroxysmal kinesigenic dyskinesias, warranting further investigations to understand the pathogenesis of this disorder.
Figures
Comment in
-
Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesia.Mov Disord. 2012 May;27(6):707. doi: 10.1002/mds.25038. Mov Disord. 2012. PMID: 22649004 No abstract available.
Similar articles
-
Mutation analysis of PRRT2 in two Chinese BFIS families and nomenclature of PRRT2 related paroxysmal diseases.Neurosci Lett. 2013 Sep 27;552:40-5. doi: 10.1016/j.neulet.2013.07.020. Epub 2013 Jul 26. Neurosci Lett. 2013. PMID: 23896529
-
The clinical and genetic heterogeneity of paroxysmal dyskinesias.Brain. 2015 Dec;138(Pt 12):3567-80. doi: 10.1093/brain/awv310. Epub 2015 Nov 23. Brain. 2015. PMID: 26598494 Free PMC article. Review.
-
Clinical and polygraphic study of familial paroxysmal kinesigenic dyskinesia with PRRT2 mutation.Epileptic Disord. 2013 Jun;15(2):123-7. doi: 10.1684/epd.2013.0569. Epileptic Disord. 2013. PMID: 23771590
-
PRRT2 mutations in a cohort of Chinese families with paroxysmal kinesigenic dyskinesia and genotype-phenotype correlation reanalysis in literatures.Int J Neurosci. 2018 Aug;128(8):751-760. doi: 10.1080/00207454.2017.1418345. Epub 2018 Jan 7. Int J Neurosci. 2018. PMID: 29285950 Review.
-
Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia.Nat Genet. 2011 Nov 20;43(12):1252-5. doi: 10.1038/ng.1008. Nat Genet. 2011. PMID: 22101681
Cited by
-
Deciphering the Role of Rapidly Evolving Conserved Elements in Primate Brain Development and Exploring Their Potential Involvement in Alzheimer's Disease.Mol Biol Evol. 2024 Jan 3;41(1):msae001. doi: 10.1093/molbev/msae001. Mol Biol Evol. 2024. PMID: 38175672 Free PMC article.
-
Clinical and genetic analyses of 150 patients with paroxysmal kinesigenic dyskinesia.J Neurol. 2022 Sep;269(9):4717-4728. doi: 10.1007/s00415-022-11103-0. Epub 2022 Apr 15. J Neurol. 2022. PMID: 35428900
-
Heterozygous RHO p.R135W missense mutation in a large Han-Chinese family with retinitis pigmentosa and different refractive errors.Biosci Rep. 2019 Jul 12;39(7):BSR20182198. doi: 10.1042/BSR20182198. Print 2019 Jul 31. Biosci Rep. 2019. PMID: 31239368 Free PMC article.
-
Comprehensive Exonic Sequencing of Hemiplegic Migraine-Related Genes in a Cohort of Suspected Probands Identifies Known and Potential Pathogenic Variants.Cells. 2020 Oct 28;9(11):2368. doi: 10.3390/cells9112368. Cells. 2020. PMID: 33126486 Free PMC article.
-
Paroxysmal Genetic Movement Disorders and Epilepsy.Front Neurol. 2021 Mar 23;12:648031. doi: 10.3389/fneur.2021.648031. eCollection 2021. Front Neurol. 2021. PMID: 33833732 Free PMC article. Review.
References
-
- Bennett LB, Roach ES, Bowcock AM. A locus for paroxysmal kinesigenic dyskinesia maps to human chromosome 16. Neurology. 2000;54:125–30. - PubMed
-
- Blakeley J, Jankovic J. Secondary paroxysmal dyskinesias. Mov Disord. 2002;17:726–34. - PubMed
-
- Bruno MK, Hallett M, Gwinn-Hardy K, Sorensen B, Considine E, Tucker S, et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology. 2004;63:2280–7. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
