

The structure of an unconventional HD-GYP protein from Bdellovibrio reveals the roles of conserved residues in this class of cyclic-di-GMP phosphodiesterases
- PMID: 21990613
- PMCID: PMC3188283
- DOI: 10.1128/mBio.00163-11
The structure of an unconventional HD-GYP protein from Bdellovibrio reveals the roles of conserved residues in this class of cyclic-di-GMP phosphodiesterases
Abstract
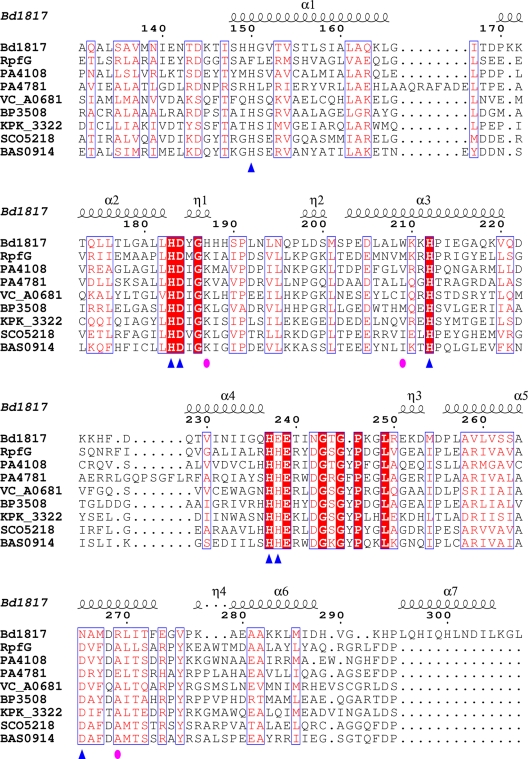
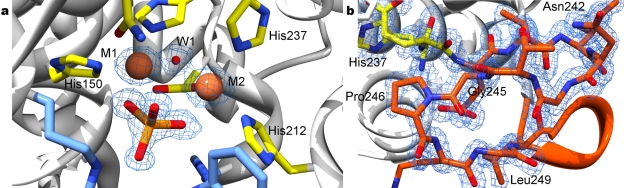
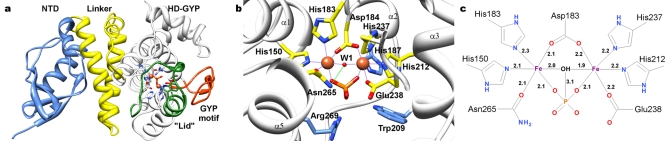
Cyclic-di-GMP is a near-ubiquitous bacterial second messenger that is important in localized signal transmission during the control of various processes, including virulence and switching between planktonic and biofilm-based lifestyles. Cyclic-di-GMP is synthesized by GGDEF diguanylate cyclases and hydrolyzed by EAL or HD-GYP phosphodiesterases, with each functional domain often appended to distinct sensory modules. HD-GYP domain proteins have resisted structural analysis, but here we present the first structural representative of this family (1.28 Å), obtained using the unusual Bd1817 HD-GYP protein from the predatory bacterium Bdellovibrio bacteriovorus. Bd1817 lacks the active-site tyrosine present in most HD-GYP family members yet remains an excellent model of their features, sharing 48% sequence similarity with the archetype RpfG. The protein structure is highly modular and thus provides a basis for delineating domain boundaries in other stimulus-dependent homologues. Conserved residues in the HD-GYP family cluster around a binuclear metal center, which is observed complexed to a molecule of phosphate, providing information on the mode of hydroxide ion attack on substrate. The fold and active site of the HD-GYP domain are different from those of EAL proteins, and restricted access to the active-site cleft is indicative of a different mode of activity regulation. The region encompassing the GYP motif has a novel conformation and is surface exposed and available for complexation with binding partners, including GGDEF proteins.
Importance: It is becoming apparent that many bacteria use the signaling molecule cyclic-di-GMP to regulate a variety of processes, most notably, transitions between motility and sessility. Importantly, this regulation is central to several traits implicated in chronic disease (adhesion, biofilm formation, and virulence gene expression). The mechanisms of cyclic-di-GMP synthesis via GGDEF enzymes and hydrolysis via EAL enzymes have been suggested by the analysis of several crystal structures, but no information has been available to date for the unrelated HD-GYP class of hydrolases. Here we present the multidomain structure of an unusual member of the HD-GYP family from the predatory bacterium Bdellovibrio bacteriovorus and detail the features that distinguish it from the wider structural family of general HD fold hydrolases. The structure reveals how a binuclear iron center is formed from several conserved residues and provides a basis for understanding HD-GYP family sequence requirements for c-di-GMP hydrolysis.
Figures
Similar articles
-
Crystal structure of an HD-GYP domain cyclic-di-GMP phosphodiesterase reveals an enzyme with a novel trinuclear catalytic iron centre.Mol Microbiol. 2014 Jan;91(1):26-38. doi: 10.1111/mmi.12447. Epub 2013 Nov 24. Mol Microbiol. 2014. PMID: 24176013 Free PMC article.
-
A systematic analysis of the in vitro and in vivo functions of the HD-GYP domain proteins of Vibrio cholerae.BMC Microbiol. 2014 Oct 25;14:272. doi: 10.1186/s12866-014-0272-9. BMC Microbiol. 2014. PMID: 25343965 Free PMC article.
-
Finally! The structural secrets of a HD-GYP phosphodiesterase revealed.Mol Microbiol. 2014 Jan;91(1):1-5. doi: 10.1111/mmi.12463. Epub 2013 Dec 1. Mol Microbiol. 2014. PMID: 24236493
-
Sequence Conservation, Domain Architectures, and Phylogenetic Distribution of the HD-GYP Type c-di-GMP Phosphodiesterases.J Bacteriol. 2022 Apr 19;204(4):e0056121. doi: 10.1128/jb.00561-21. Epub 2021 Dec 20. J Bacteriol. 2022. PMID: 34928179 Free PMC article. Review.
-
The HD-GYP domain, cyclic di-GMP signaling, and bacterial virulence to plants.Mol Plant Microbe Interact. 2006 Dec;19(12):1378-84. doi: 10.1094/MPMI-19-1378. Mol Plant Microbe Interact. 2006. PMID: 17153922 Review.
Cited by
-
Diffusible Signaling Factor, a Quorum-Sensing Molecule, Interferes with and Is Toxic Towards Bdellovibrio bacteriovorus 109J.Microb Ecol. 2021 Feb;81(2):347-356. doi: 10.1007/s00248-020-01585-8. Epub 2020 Sep 6. Microb Ecol. 2021. PMID: 32892232
-
Advances in cellular and molecular predatory biology of Bdellovibrio bacteriovorus six decades after discovery.Front Microbiol. 2023 May 15;14:1168709. doi: 10.3389/fmicb.2023.1168709. eCollection 2023. Front Microbiol. 2023. PMID: 37256055 Free PMC article. Review.
-
The Characterization of Escherichia coli CpdB as a Recombinant Protein Reveals that, besides Having the Expected 3´-Nucleotidase and 2´,3´-Cyclic Mononucleotide Phosphodiesterase Activities, It Is Also Active as Cyclic Dinucleotide Phosphodiesterase.PLoS One. 2016 Jun 13;11(6):e0157308. doi: 10.1371/journal.pone.0157308. eCollection 2016. PLoS One. 2016. PMID: 27294396 Free PMC article.
-
Bacterial rotary export ATPases are allosterically regulated by the nucleotide second messenger cyclic-di-GMP.J Biol Chem. 2015 Oct 2;290(40):24470-83. doi: 10.1074/jbc.M115.661439. Epub 2015 Aug 11. J Biol Chem. 2015. PMID: 26265469 Free PMC article.
-
c-di-GMP Arms an Anti-σ to Control Progression of Multicellular Differentiation in Streptomyces.Mol Cell. 2020 Feb 6;77(3):586-599.e6. doi: 10.1016/j.molcel.2019.11.006. Epub 2019 Dec 3. Mol Cell. 2020. PMID: 31810759 Free PMC article.
References
-
- Römling U, Gomelsky M, Galperin MY. 2005. C-di-GMP: the dawning of a novel bacterial signalling system. Mol. Microbiol. 57:629–639 - PubMed
-
- Schirmer T, Jenal U. 2009. Structural and mechanistic determinants of c-di-GMP signalling. Nat. Rev. Microbiol. 7:724–735 - PubMed
-
- Ross P, et al. 1987. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature 325:279–281 - PubMed
-
- Amikam D, Galperin MY. 2006. PilZ domain is part of the bacterial c-di-GMP binding protein. Bioinformatics 22:3–6 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
