

Reversible infantile respiratory chain deficiency is a unique, genetically heterogenous mitochondrial disease
- PMID: 21931168
- PMCID: PMC4562368
- DOI: 10.1136/jmg.2011.089995
Reversible infantile respiratory chain deficiency is a unique, genetically heterogenous mitochondrial disease
Abstract
Objectives: Homoplasmic maternally inherited, m.14674T>C or m. 14674T>G mt-tRNA(Glu) mutations have recently been identified in reversible infantile cytochrome c oxidase deficiency (or 'benign COX deficiency'). This study sought other genetic defects that may give rise to similar presentations.
Patients: Eight patients from seven families with clinicopathological features of infantile reversible cytochrome c oxidase deficiency were investigated.
Methods: The study reviewed the diagnostic features and performed molecular genetic analyses of mitochondrial DNA and nuclear encoded candidate genes.
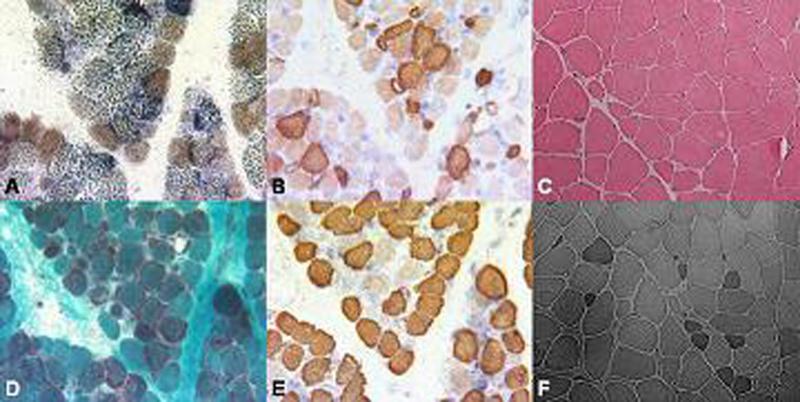
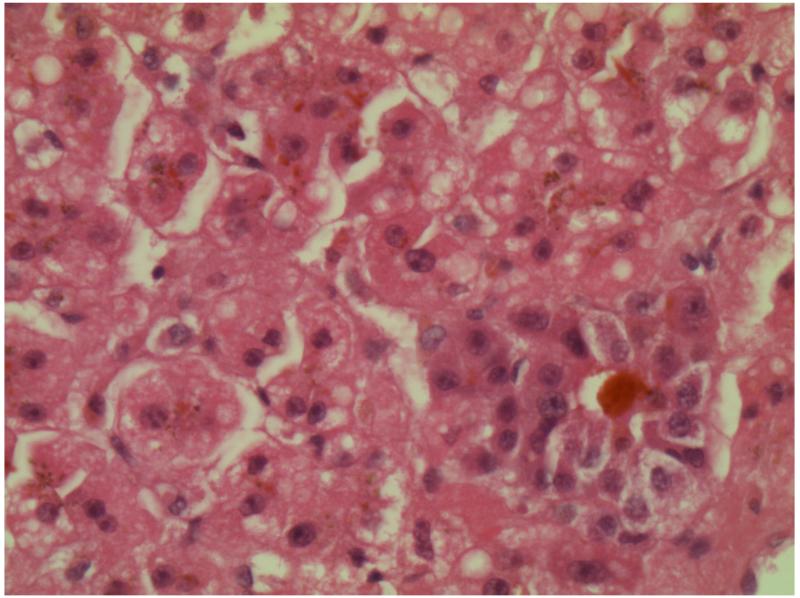
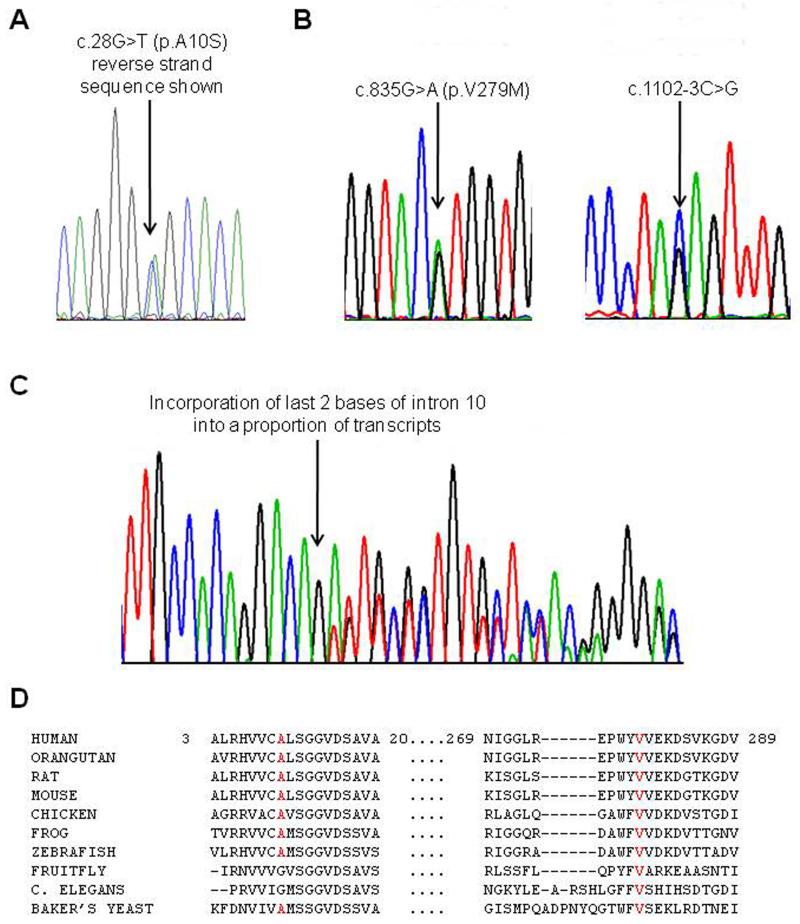
Results: Patients presented with subacute onset of profound hypotonia, feeding difficulties and lactic acidosis within the first months of life. Although recovery was remarkable, a mild myopathy persisted into adulthood. Histopathological findings in muscle included increased lipid and/or glycogen content, ragged-red and COX negative fibres. Biochemical studies suggested more generalised abnormalities than pure COX deficiency. Clinical improvement was reflected by normalisation of lactic acidosis and histopathological abnormalities. The m.14674T>C mt-tRNA(Glu) mutation was identified in four families, but none had the m. 14674T>G mutation. Furthermore, in two families pathogenic mutations were also found in the nuclear TRMU gene which has not previously been associated with this phenotype. In one family, the genetic aetiology still remains unknown.
Conclusions: Benign COX deficiency is better described as 'reversible infantile respiratory chain deficiency'. It is genetically heterogeneous, and patients not carrying the m.14674T>C or T>G mt-tRNA(Glu) mutations may have mutations in the TRMU gene. Diagnosing this disorder at the molecular level is a significant advance for paediatric neurologists and intensive care paediatricians, enabling them to select children with an excellent prognosis for continuing respiratory support from those with severe mitochondrial presentation in infancy.
Figures
Similar articles
-
Expression pattern of mitochondrial respiratory chain enzymes in skeletal muscle of patients with mitochondrial myopathy associated with the homoplasmic m.14674T>C variant.Brain Pathol. 2022 Jul;32(4):e13038. doi: 10.1111/bpa.13038. Epub 2021 Nov 21. Brain Pathol. 2022. PMID: 34806237 Free PMC article.
-
Molecular basis of infantile reversible cytochrome c oxidase deficiency myopathy.Brain. 2009 Nov;132(Pt 11):3165-74. doi: 10.1093/brain/awp221. Epub 2009 Aug 31. Brain. 2009. PMID: 19720722 Free PMC article.
-
Muscle fat replacement and modified ragged red fibers in two patients with reversible infantile respiratory chain deficiency.Neuromuscul Disord. 2021 Jun;31(6):551-557. doi: 10.1016/j.nmd.2021.02.017. Epub 2021 Feb 21. Neuromuscul Disord. 2021. PMID: 33832841
-
Defects of the mitochondrial respiratory chain complexes in three pediatric cases with hypotonia and cardiac involvement.J Neurol Sci. 1992 Mar;108(1):105-13. doi: 10.1016/0022-510x(92)90195-q. J Neurol Sci. 1992. PMID: 1320661 Review.
-
Infantile cardioencephalopathy due to a COX15 gene defect: report and review.Am J Med Genet A. 2011 Apr;155A(4):840-4. doi: 10.1002/ajmg.a.33881. Epub 2011 Mar 15. Am J Med Genet A. 2011. PMID: 21412973 Review.
Cited by
-
Pathogenic mitochondrial tRNA point mutations: nine novel mutations affirm their importance as a cause of mitochondrial disease.Hum Mutat. 2013 Sep;34(9):1260-8. doi: 10.1002/humu.22358. Hum Mutat. 2013. PMID: 23696415 Free PMC article.
-
Pathological mutations promote proteolysis of mitochondrial tRNA-specific 2-thiouridylase 1 (MTU1) via mitochondrial caseinolytic peptidase (CLPP).Nucleic Acids Res. 2024 Feb 9;52(3):1341-1358. doi: 10.1093/nar/gkad1197. Nucleic Acids Res. 2024. PMID: 38113276 Free PMC article.
-
Genetic polymorphisms associated with adverse pregnancy outcomes in nulliparas.Sci Rep. 2024 May 7;14(1):10514. doi: 10.1038/s41598-024-61218-9. Sci Rep. 2024. PMID: 38714721 Free PMC article.
-
Whole-exome sequencing identified novel compound heterozygous variants in a Chinese neonate with liver failure and review of literature.Mol Genet Genomic Med. 2020 Dec;8(12):e1515. doi: 10.1002/mgg3.1515. Epub 2020 Nov 18. Mol Genet Genomic Med. 2020. PMID: 33205917 Free PMC article. Review.
-
Leigh syndrome associated with TRMU gene mutations.Mol Genet Metab Rep. 2020 Dec 15;26:100690. doi: 10.1016/j.ymgmr.2020.100690. eCollection 2021 Mar. Mol Genet Metab Rep. 2020. PMID: 33365252 Free PMC article.
References
-
- DiMauro S, Nicholson JF, Hays AP, et al. Benign infantile mitochondrial myopathy due to reversible cytochrome c oxidase deficiency. Trans Am Neurol Assoc. 1981;106:205–7. - PubMed
-
- DiMauro S, Nicholson JF, Hays AP, et al. Benign infantile mitochondrial myopathy due to reversible cytochrome c oxidase deficiency. Ann Neurol. 1983;14(2):226–34. - PubMed
-
- Roodhooft AM, Van Acker KJ, Martin JJ, et al. Benign mitochondrial myopathy with deficiency of NADHCoQ reductase and cytochrome c oxidase. Neuropediatrics. 1986;17:221–26. - PubMed
-
- Zeviani M, Peterson P, Servidei S, et al. Benign reversible muscle cytochrome c oxidase deficiency: a second case. Neurology. 1987;37(1):64–7. - PubMed
-
- Nonaka I, Koga Y, Shikura K, et al. Muscle pathology in cytochrome c oxidase deficiency. Acta Neuropathol. 1988;77:152–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases