

Genotype-phenotype correlation in primary carnitine deficiency
- PMID: 21922592
- PMCID: PMC3240685
- DOI: 10.1002/humu.21607
Genotype-phenotype correlation in primary carnitine deficiency
Abstract
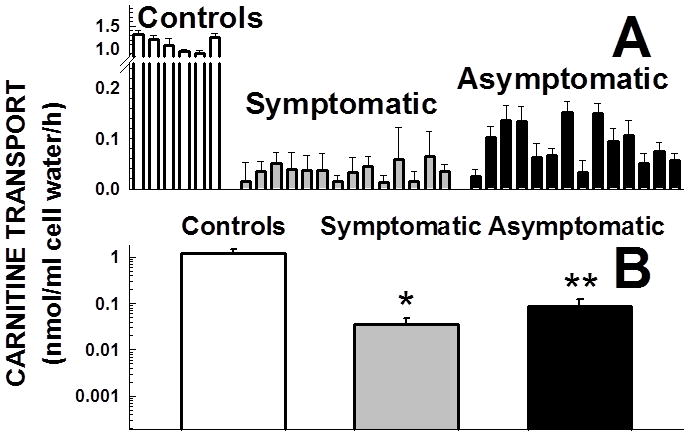
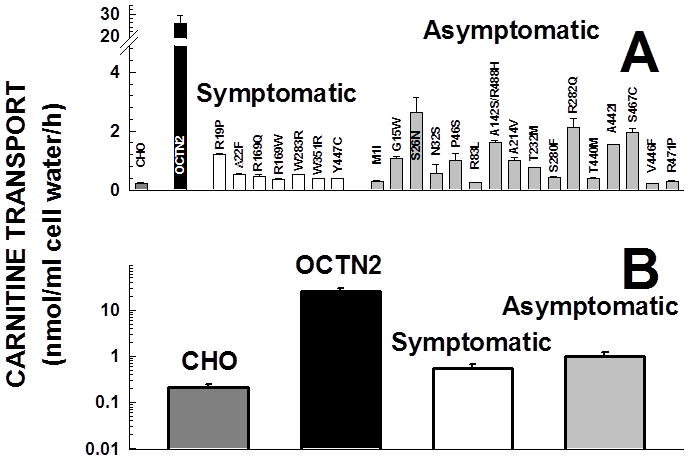
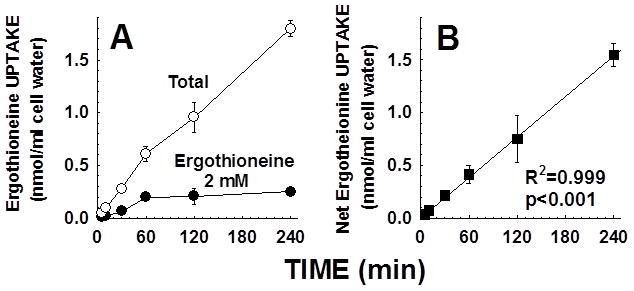
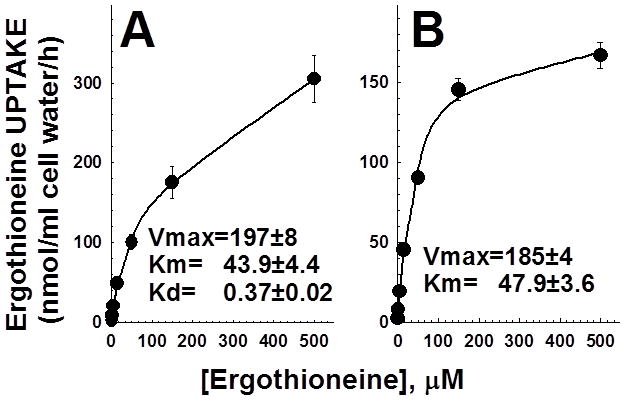
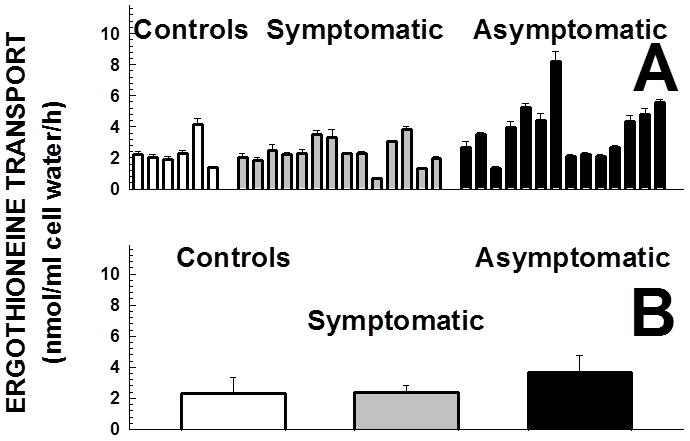
Primary carnitine deficiency is caused by defective OCTN2 carnitine transporters encoded by the SLC22A5 gene. Lack of carnitine impairs fatty acid oxidation resulting in hypoketotic hypoglycemia, hepatic encephalopathy, skeletal and cardiac myopathy. Recently, asymptomatic mothers with primary carnitine deficiency were identified by low carnitine levels in their infant by newborn screening. Here, we evaluate mutations in the SLC22A5 gene and carnitine transport in fibroblasts from symptomatic patients and asymptomatic women. Carnitine transport was significantly reduced in fibroblasts obtained from all patients with primary carnitine deficiency, but was significantly higher in the asymptomatic women's than in the symptomatic patients' fibroblasts (P < 0.01). By contrast, ergothioneine transport (a selective substrate of the OCTN1 transporter, tested here as a control) was similar in cells from controls and patients with carnitine deficiency. DNA sequencing indicated an increased frequency of nonsense mutations in symptomatic patients (P < 0.001). Expression of the missense mutations in Chinese hamster ovary (CHO) cells indicated that many mutations retained residual carnitine transport activity, with no difference in the average activity of missense mutations identified in symptomatic versus asymptomatic patients. These results indicate that cells from asymptomatic women have on average higher levels of residual carnitine transport activity as compared to that of symptomatic patients due to the presence of at least one missense mutation.
© 2011 Wiley Periodicals, Inc.
Figures
Comment in
-
Carnitine uptake defect (primary carnitine deficiency): risk in genotype-phenotype correlation.Hum Mutat. 2013 Apr;34(4):655. doi: 10.1002/humu.22286. Hum Mutat. 2013. PMID: 23520115 No abstract available.
Similar articles
-
Functional and molecular studies in primary carnitine deficiency.Hum Mutat. 2017 Dec;38(12):1684-1699. doi: 10.1002/humu.23315. Epub 2017 Sep 14. Hum Mutat. 2017. PMID: 28841266 Free PMC article.
-
Analysis of genetic mutations in Chinese patients with systemic primary carnitine deficiency.Eur J Med Genet. 2014 Oct;57(10):571-5. doi: 10.1016/j.ejmg.2014.08.001. Epub 2014 Aug 13. Eur J Med Genet. 2014. PMID: 25132046
-
Glycosylation of the OCTN2 carnitine transporter: study of natural mutations identified in patients with primary carnitine deficiency.Biochim Biophys Acta. 2011 Mar;1812(3):312-20. doi: 10.1016/j.bbadis.2010.11.007. Epub 2010 Nov 29. Biochim Biophys Acta. 2011. PMID: 21126579 Free PMC article.
-
Carnitine transport and fatty acid oxidation.Biochim Biophys Acta. 2016 Oct;1863(10):2422-35. doi: 10.1016/j.bbamcr.2016.01.023. Epub 2016 Jan 29. Biochim Biophys Acta. 2016. PMID: 26828774 Free PMC article. Review.
-
[Carnitine - mitochondria and beyond].Postepy Biochem. 2016;62(2):85-93. Postepy Biochem. 2016. PMID: 28132459 Review. Polish.
Cited by
-
Rechallenge of immune checkpoint inhibitors in a case with adverse events inducing myasthenia gravis.J Immunother Cancer. 2022 Nov;10(11):e005970. doi: 10.1136/jitc-2022-005970. J Immunother Cancer. 2022. PMID: 36450378 Free PMC article.
-
Pathophysiology of fatty acid oxidation disorders and resultant phenotypic variability.J Inherit Metab Dis. 2013 Jul;36(4):645-58. doi: 10.1007/s10545-013-9611-5. Epub 2013 May 15. J Inherit Metab Dis. 2013. PMID: 23674167 Free PMC article. Review.
-
A Retrospective Analysis of Clinically Focused Exome Sequencing Results of 372 Infants with Suspected Monogenic Disorders in China.Pharmgenomics Pers Med. 2023 Feb 2;16:81-97. doi: 10.2147/PGPM.S387767. eCollection 2023. Pharmgenomics Pers Med. 2023. PMID: 36755623 Free PMC article.
-
Clinical characteristics of primary carnitine deficiency: A structured review using a case-by-case approach.J Inherit Metab Dis. 2022 May;45(3):386-405. doi: 10.1002/jimd.12475. Epub 2022 Feb 3. J Inherit Metab Dis. 2022. PMID: 34997761 Free PMC article. Review.
-
Organic Cation Transporters in Human Physiology, Pharmacology, and Toxicology.Int J Mol Sci. 2020 Oct 24;21(21):7890. doi: 10.3390/ijms21217890. Int J Mol Sci. 2020. PMID: 33114309 Free PMC article. Review.
References
-
- Amat Di San Filippo C, Longo N. Tyrosine Residues Affecting Sodium Stimulation of Carnitine Transport in the OCTN2 Carnitine/Organic Cation Transporter. J Biol Chem. 2004;279:7247–53. - PubMed
-
- Amat di San Filippo C, Pasquali M, Longo N. Pharmacological rescue of carnitine transport in primary carnitine deficiency. Hum Mutat. 2006;27:513–23. - PubMed
-
- Amat di San Filippo C, Wang Y, Longo N. Functional domains in the carnitine transporter OCTN2, defective in primary carnitine deficiency. J Biol Chem. 2003;278:47776–84. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
