

Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS)
- PMID: 21282379
- PMCID: PMC3058571
- DOI: 10.1084/jem.20102049
Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS)
Abstract
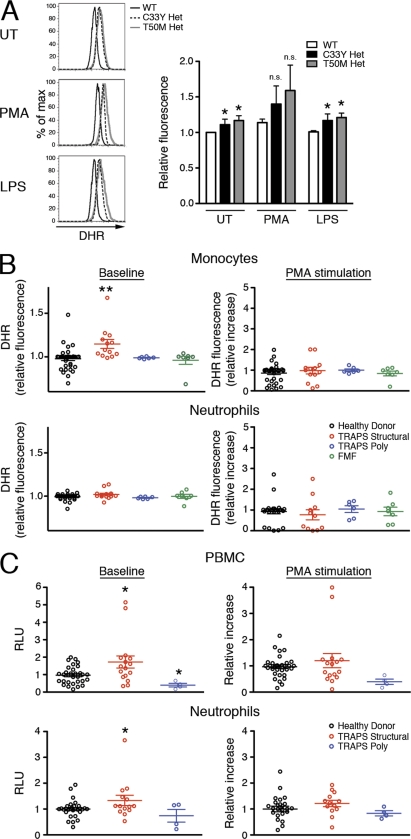
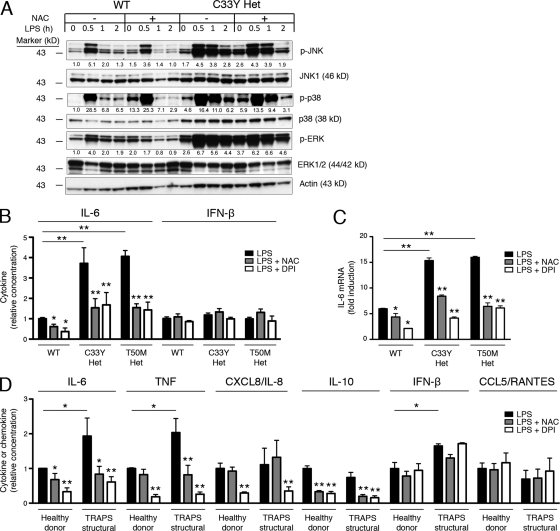
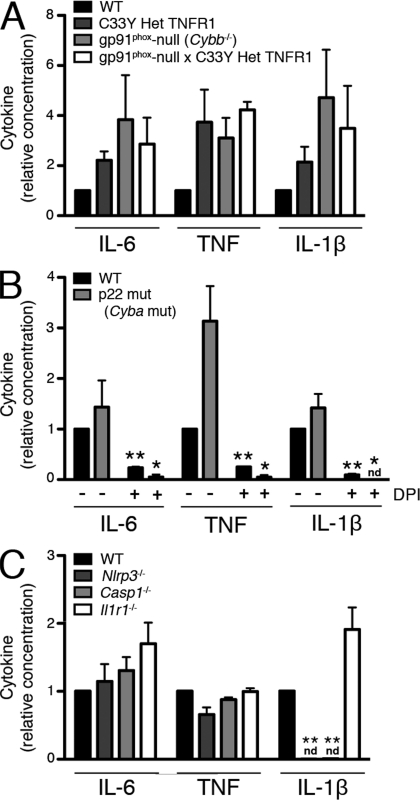
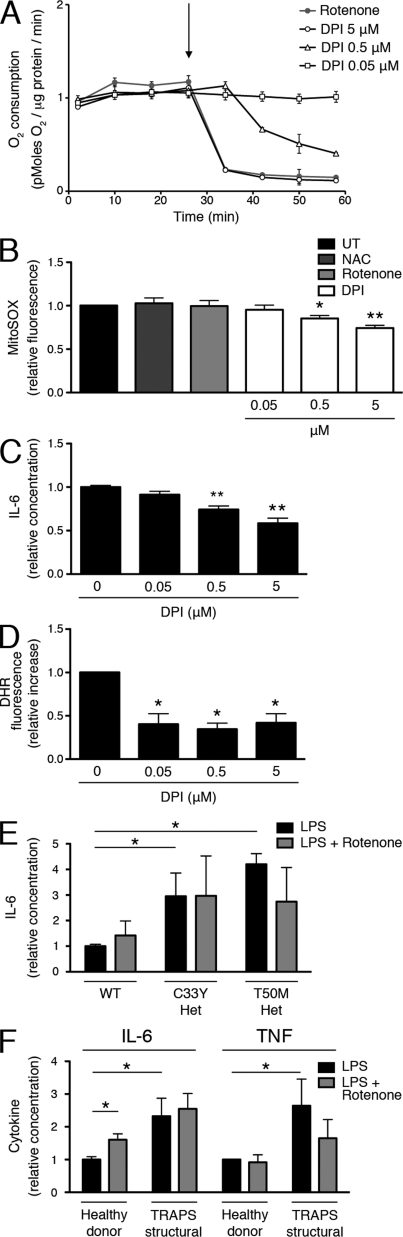
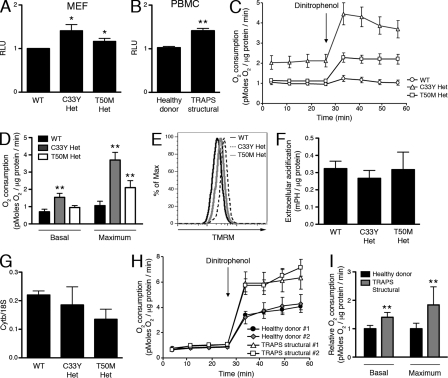
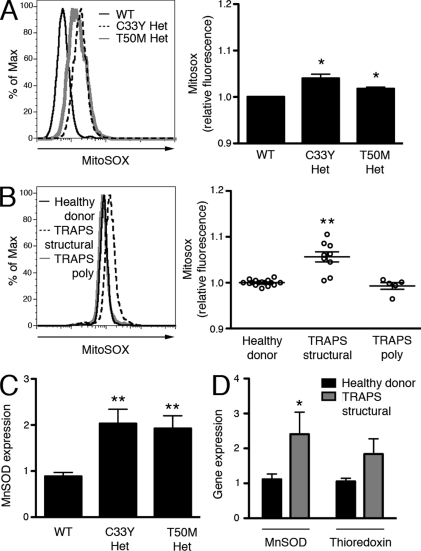
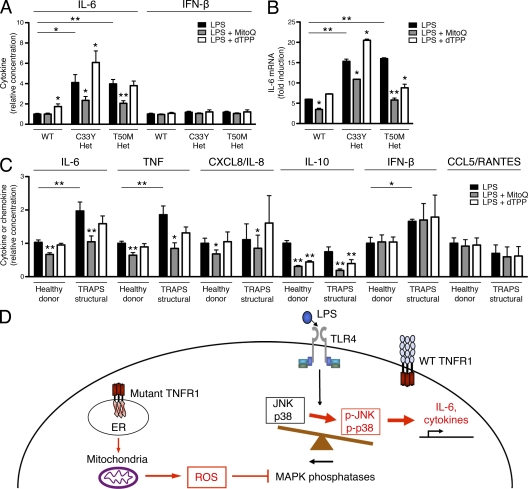
Reactive oxygen species (ROS) have an established role in inflammation and host defense, as they kill intracellular bacteria and have been shown to activate the NLRP3 inflammasome. Here, we find that ROS generated by mitochondrial respiration are important for normal lipopolysaccharide (LPS)-driven production of several proinflammatory cytokines and for the enhanced responsiveness to LPS seen in cells from patients with tumor necrosis factor receptor-associated periodic syndrome (TRAPS), an autoinflammatory disorder caused by missense mutations in the type 1 TNF receptor (TNFR1). We find elevated baseline ROS in both mouse embryonic fibroblasts and human immune cells harboring TRAPS-associated TNFR1 mutations. A variety of antioxidants dampen LPS-induced MAPK phosphorylation and inflammatory cytokine production. However, gp91(phox) and p22(phox) reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits are dispensable for inflammatory cytokine production, indicating that NADPH oxidases are not the source of proinflammatory ROS. TNFR1 mutant cells exhibit altered mitochondrial function with enhanced oxidative capacity and mitochondrial ROS generation, and pharmacological blockade of mitochondrial ROS efficiently reduces inflammatory cytokine production after LPS stimulation in cells from TRAPS patients and healthy controls. These findings suggest that mitochondrial ROS may be a novel therapeutic target for TRAPS and other inflammatory diseases.
Figures
Comment in
-
Inflammation: ROSy outlook for TRAPS patients.Nat Rev Immunol. 2011 Mar;11(3):162. doi: 10.1038/nri2943. Nat Rev Immunol. 2011. PMID: 21456316 No abstract available.
-
Inflammation: insights into the role of mitochondrial reactive oxygen species in the pathogenesis of TRAPS.Nat Rev Rheumatol. 2011 May;7(5):254. doi: 10.1038/nrrheum.2011.47. Epub 2011 Apr 5. Nat Rev Rheumatol. 2011. PMID: 21468146 No abstract available.
Similar articles
-
Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome.Proc Natl Acad Sci U S A. 2010 May 25;107(21):9801-6. doi: 10.1073/pnas.0914118107. Epub 2010 May 10. Proc Natl Acad Sci U S A. 2010. PMID: 20457915 Free PMC article.
-
Mitochondrial Reactive Oxygen Species Participate in Signaling Triggered by Heme in Macrophages and upon Hemolysis.J Immunol. 2020 Nov 15;205(10):2795-2805. doi: 10.4049/jimmunol.1900886. Epub 2020 Oct 9. J Immunol. 2020. PMID: 33037139
-
TRAPS mutations in Tnfrsf1a decrease the responsiveness to TNFα via reduced cell surface expression of TNFR1.Front Immunol. 2022 Jul 22;13:926175. doi: 10.3389/fimmu.2022.926175. eCollection 2022. Front Immunol. 2022. PMID: 35936010 Free PMC article.
-
Mitochondrial reactive oxygen species drive proinflammatory cytokine production.J Exp Med. 2011 Mar 14;208(3):417-20. doi: 10.1084/jem.20110367. Epub 2011 Feb 28. J Exp Med. 2011. PMID: 21357740 Free PMC article. Review.
-
[Progress in classification and treatment for TNF receptor-associated periodic syndrome].Nihon Rinsho Meneki Gakkai Kaishi. 2011;34(5):361-8. doi: 10.2177/jsci.34.361. Nihon Rinsho Meneki Gakkai Kaishi. 2011. PMID: 22041423 Review. Japanese.
Cited by
-
Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF.Oxid Med Cell Longev. 2015;2015:610813. doi: 10.1155/2015/610813. Epub 2015 Mar 5. Oxid Med Cell Longev. 2015. PMID: 25834699 Free PMC article. Review.
-
Mitochondria-derived hydrogen peroxide selectively enhances T cell receptor-initiated signal transduction.J Biol Chem. 2013 Sep 6;288(36):26246-26255. doi: 10.1074/jbc.M113.476895. Epub 2013 Jul 23. J Biol Chem. 2013. PMID: 23880762 Free PMC article.
-
A single gene of a commensal microbe affects host susceptibility to enteric infection.Nat Commun. 2016 May 13;7:11606. doi: 10.1038/ncomms11606. Nat Commun. 2016. PMID: 27173141 Free PMC article.
-
Chitohexaose activates macrophages by alternate pathway through TLR4 and blocks endotoxemia.PLoS Pathog. 2012;8(5):e1002717. doi: 10.1371/journal.ppat.1002717. Epub 2012 May 24. PLoS Pathog. 2012. PMID: 22654663 Free PMC article.
-
Involvement of the same TNFR1 residue in mendelian and multifactorial inflammatory disorders.PLoS One. 2013 Jul 24;8(7):e69757. doi: 10.1371/journal.pone.0069757. Print 2013. PLoS One. 2013. PMID: 23894535 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
