

Integrated analysis of clinical signs and literature data for the diagnosis and therapy of a previously undescribed 6p21.3 deletion syndrome
- PMID: 21119708
- PMCID: PMC3025798
- DOI: 10.1038/ejhg.2010.172
Integrated analysis of clinical signs and literature data for the diagnosis and therapy of a previously undescribed 6p21.3 deletion syndrome
Abstract
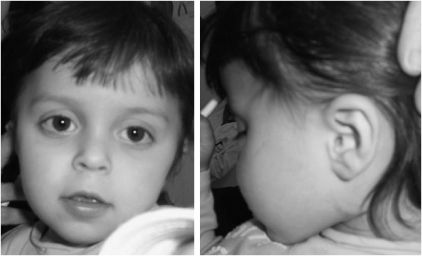
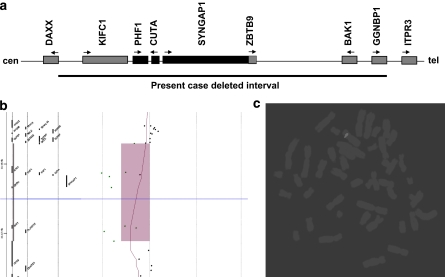
A de novo 0.3 Mb deletion on 6p21.3 was detected by array-comparative genomic hybridization in a girl with mental retardation, drug-resistant seizures, facial dysmorphisms, gut malrotation and abnormal pancreas segmentation. Consistent with phenotypic manifestations is haploinsufficiency of SYNGAP1, which was recently demonstrated to cause non-syndromic mental retardation, and of the flanking genes CuTA, a likely modulator of the processing and trafficking of secretory proteins in the human brain, and hPHF1, involved in HOX gene silencing. Mutations of both CuTA and hPHF1 were never reported as causative of human diseases. Similarly, the present syndromic condition was not previously described and it can be regarded as a human model confirming the suggested biological properties of the genes included in the deletion interval. In addition, experimental evidence that SYNGAP1 and CuTA are involved in the secretory pathway in neurons, through glutamate and acetylcholinesterase signalling, prompted us to consider modulation of the glutamate pathway as target of a therapeutic strategy for seizure control.
Figures
Similar articles
-
6p21.3 microdeletion involving the SYNGAP1 gene in a patient with intellectual disability, seizures, and severe speech impairment.Am J Med Genet A. 2013 Jul;161A(7):1682-5. doi: 10.1002/ajmg.a.35930. Epub 2013 May 17. Am J Med Genet A. 2013. PMID: 23687080
-
De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability.Am J Med Genet A. 2015 Oct;167A(10):2231-7. doi: 10.1002/ajmg.a.37189. Epub 2015 Jun 15. Am J Med Genet A. 2015. PMID: 26079862 Free PMC article.
-
A novel de novo microdeletion spanning the SYNGAP1 gene on the short arm of chromosome 6 associated with mental retardation.Am J Med Genet A. 2010 Sep;152A(9):2376-8. doi: 10.1002/ajmg.a.33554. Am J Med Genet A. 2010. PMID: 20683986 No abstract available.
-
6p subtelomere deletion with congenital glaucoma, severe mental retardation, and growth impairment.Pediatr Int. 2013 Jun;55(3):376-81. doi: 10.1111/j.1442-200X.2012.03729.x. Pediatr Int. 2013. PMID: 23782370 Review.
-
De novo microdeletions of chromosome 6q14.1-q14.3 and 6q12.1-q14.1 in two patients with intellectual disability - further delineation of the 6q14 microdeletion syndrome and review of the literature.Eur J Med Genet. 2012 Aug-Sep;55(8-9):490-7. doi: 10.1016/j.ejmg.2012.03.003. Epub 2012 Apr 12. Eur J Med Genet. 2012. PMID: 22561202 Review.
Cited by
-
Identification of an individual with a SYGNAP1 pathogenic mutation in India.Mol Biol Rep. 2020 Nov;47(11):9225-9234. doi: 10.1007/s11033-020-05915-4. Epub 2020 Oct 22. Mol Biol Rep. 2020. PMID: 33090308
-
SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy.Neurology. 2019 Jan 8;92(2):e96-e107. doi: 10.1212/WNL.0000000000006729. Epub 2018 Dec 12. Neurology. 2019. PMID: 30541864 Free PMC article.
-
Case report: Off-label use of low-dose perampanel in a 25-month-old girl with a pathogenic SYNGAP1 variant.Front Neurol. 2023 Aug 17;14:1221161. doi: 10.3389/fneur.2023.1221161. eCollection 2023. Front Neurol. 2023. PMID: 37662032 Free PMC article.
-
Comprehensive behavioral analysis of heterozygous Syngap1 knockout mice.Neuropsychopharmacol Rep. 2019 Sep;39(3):223-237. doi: 10.1002/npr2.12073. Epub 2019 Jul 19. Neuropsychopharmacol Rep. 2019. PMID: 31323176 Free PMC article.
-
Transcriptional signatures of participant-derived neural progenitor cells and neurons implicate altered Wnt signaling in Phelan-McDermid syndrome and autism.Mol Autism. 2020 Jun 19;11(1):53. doi: 10.1186/s13229-020-00355-0. Mol Autism. 2020. PMID: 32560742 Free PMC article.
References
-
- Lee C, Iafrate AJ, Brothman AR. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet. 2007;39:S48–S54. - PubMed
-
- Koolen DA, Pfundt R, de Leeuw N, et al. Genomic microarrays in mental retardation: a practical workflow for diagnostic applications. Hum Mutat. 2009;30:283–292. - PubMed
-
- Purpura DP. Dendritic spine ‘dysgenesis' and mental retardation. Science. 1974;186:1126–1128. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
