

Inherited human diseases of heterotopic bone formation
- PMID: 20703219
- PMCID: PMC3551620
- DOI: 10.1038/nrrheum.2010.122
Inherited human diseases of heterotopic bone formation
Abstract
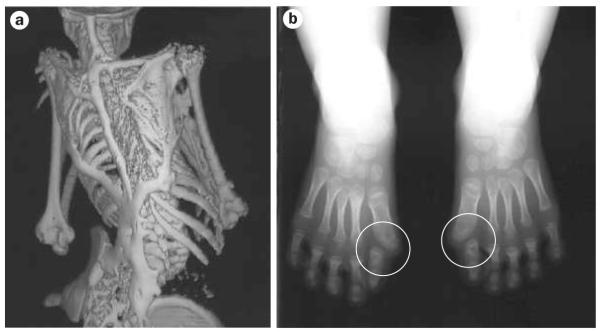
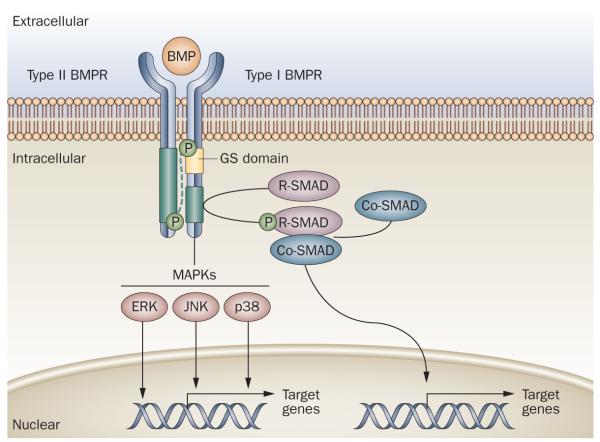
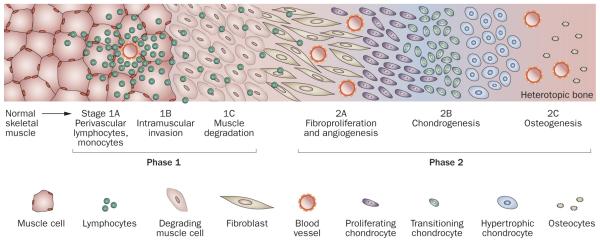
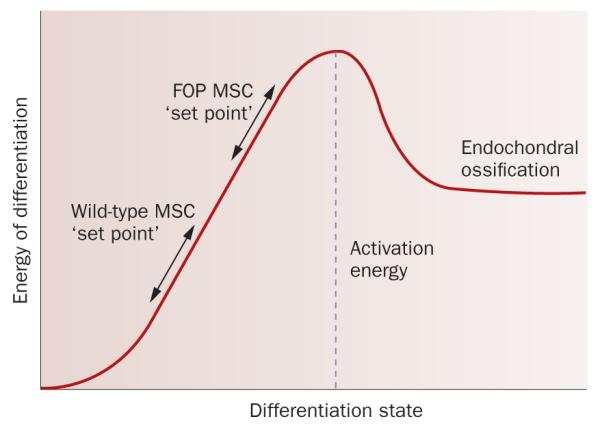
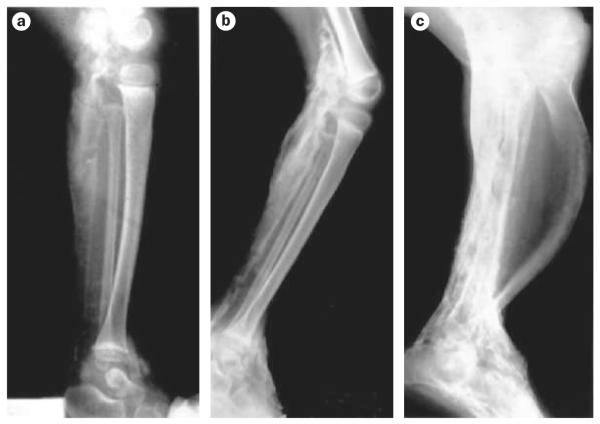
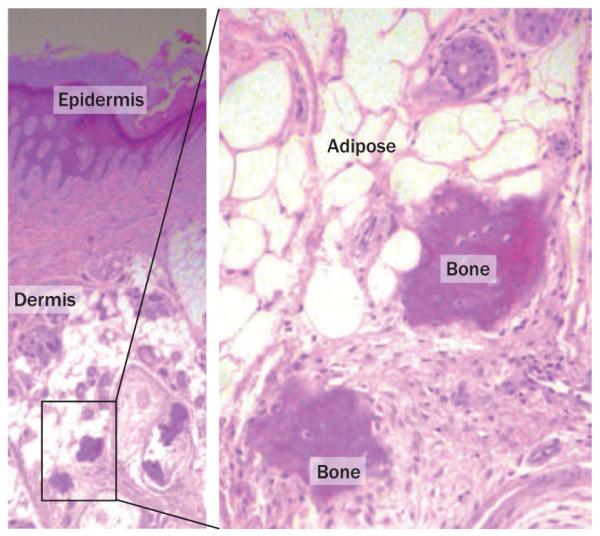
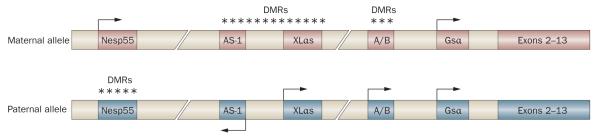
Human disorders of hereditary and nonhereditary heterotopic ossification are conditions in which osteogenesis occurs outside of the skeleton, within soft tissues of the body. The resulting extraskeletal bone is normal. The aberration lies within the mechanisms that regulate cell-fate determination, directing the inappropriate formation of cartilage or bone, or both, in tissues such as skeletal muscle and adipose tissue. Specific gene mutations have been identified in two rare inherited disorders that are clinically characterized by extensive and progressive extraskeletal bone formation-fibrodysplasia ossificans progressiva and progressive osseous heteroplasia. In fibrodysplasia ossificans progressiva, activating mutations in activin receptor type-1, a bone morphogenetic protein type I receptor, induce heterotopic endochondral ossification, which results in the development of a functional bone organ system that includes skeletal-like bone and bone marrow. In progressive osseous heteroplasia, the heterotopic ossification leads to the formation of mainly intramembranous bone tissue in response to inactivating mutations in the GNAS gene. Patients with these diseases variably show malformation of normal skeletal elements, identifying the causative genes and their associated signaling pathways as key mediators of skeletal development in addition to regulating cell-fate decisions by adult stem cells.
Figures
Similar articles
-
Role of altered signal transduction in heterotopic ossification and fibrodysplasia ossificans progressiva.Curr Osteoporos Rep. 2011 Jun;9(2):83-8. doi: 10.1007/s11914-011-0046-3. Curr Osteoporos Rep. 2011. PMID: 21340697 Free PMC article. Review.
-
Alk2 regulates early chondrogenic fate in fibrodysplasia ossificans progressiva heterotopic endochondral ossification.Stem Cells. 2014 May;32(5):1289-300. doi: 10.1002/stem.1633. Stem Cells. 2014. PMID: 24449086 Free PMC article.
-
Osteogenic induction in hereditary disorders of heterotopic ossification.Clin Orthop Relat Res. 2000 May;(374):303-16. doi: 10.1097/00003086-200005000-00028. Clin Orthop Relat Res. 2000. PMID: 10818990 Review.
-
Hematopoietic stem-cell contribution to ectopic skeletogenesis.J Bone Joint Surg Am. 2007 Feb;89(2):347-57. doi: 10.2106/JBJS.F.00472. J Bone Joint Surg Am. 2007. PMID: 17272450
-
Fibrodysplasia ossificans progressiva: a human genetic disorder of extraskeletal bone formation, or--how does one tissue become another?Wiley Interdiscip Rev Dev Biol. 2012 Jan-Feb;1(1):153-65. doi: 10.1002/wdev.9. Wiley Interdiscip Rev Dev Biol. 2012. PMID: 22408652 Free PMC article.
Cited by
-
The face signature of fibrodysplasia ossificans progressiva.Am J Med Genet A. 2012 Jun;158A(6):1368-80. doi: 10.1002/ajmg.a.35346. Epub 2012 May 11. Am J Med Genet A. 2012. PMID: 22581580 Free PMC article.
-
The role of the 3'UTR region in the regulation of the ACVR1/Alk-2 gene expression.PLoS One. 2012;7(12):e50958. doi: 10.1371/journal.pone.0050958. Epub 2012 Dec 5. PLoS One. 2012. PMID: 23227223 Free PMC article.
-
Heterotopic ossification: an unusual presentation.Case Rep Dent. 2012;2012:516717. doi: 10.1155/2012/516717. Epub 2012 Dec 31. Case Rep Dent. 2012. PMID: 23346421 Free PMC article.
-
Role of stem/progenitor cells in reparative disorders.Fibrogenesis Tissue Repair. 2012 Dec 27;5(1):20. doi: 10.1186/1755-1536-5-20. Fibrogenesis Tissue Repair. 2012. PMID: 23270300 Free PMC article.
-
Calcification of joints and arteries: second report with novel NT5E mutations and expansion of the phenotype.J Hum Genet. 2015 Oct;60(10):561-4. doi: 10.1038/jhg.2015.85. Epub 2015 Jul 16. J Hum Genet. 2015. PMID: 26178434 Clinical Trial.
References
-
- Yang Y. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Rosen CJ, editor. American Society of Bone and Mineral Research; Washington, DC: 2008. pp. 2–10.
-
- McCarthy EF, Sundaram M. Heterotopic ossification: a review. Skeletal Radiol. 2005;34:609–619. - PubMed
-
- Pignolo RJ, Foley KL. Nonhereditary heterotopic ossification. Implications for injury, arthropy, and aging. Clin. Rev. Bone Miner. Metab. 2005;3:261–266.
-
- Forsberg JA, et al. Heterotopic ossification in high-energy wartime extremity injuries: prevalence and risk factors. J. Bone Joint Surg. Am. 2009;91:1084–1091. - PubMed
-
- Neal B, Gray H, MacMahon S, Dunn L. Incidence of heterotopic bone formation after major hip surgery. ANZ J. Surg. 2002;72:808–821. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
