

Joubert Syndrome and related disorders
- PMID: 20615230
- PMCID: PMC2913941
- DOI: 10.1186/1750-1172-5-20
Joubert Syndrome and related disorders
Abstract
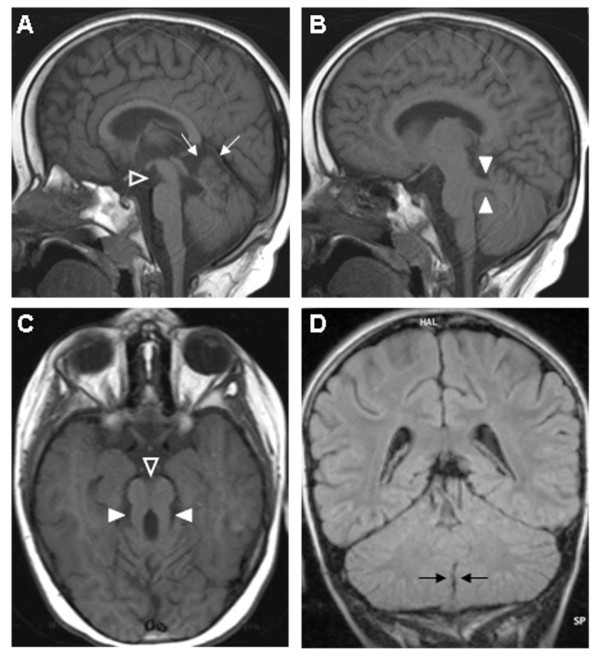
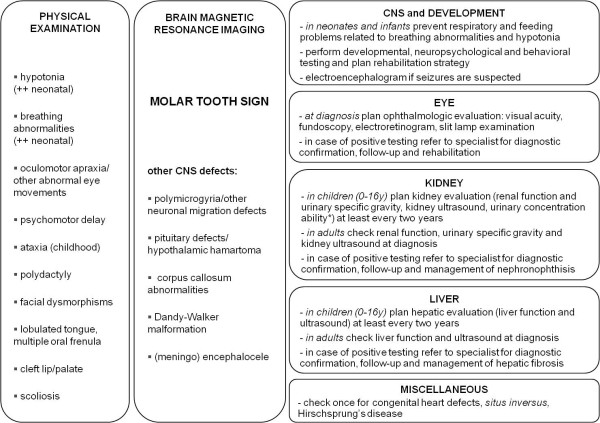
Joubert syndrome (JS) and related disorders (JSRD) are a group of developmental delay/multiple congenital anomalies syndromes in which the obligatory hallmark is the molar tooth sign (MTS), a complex midbrain-hindbrain malformation visible on brain imaging, first recognized in JS. Estimates of the incidence of JSRD range between 1/80,000 and 1/100,000 live births, although these figures may represent an underestimate. The neurological features of JSRD include hypotonia, ataxia, developmental delay, intellectual disability, abnormal eye movements, and neonatal breathing dysregulation. These may be associated with multiorgan involvement, mainly retinal dystrophy, nephronophthisis, hepatic fibrosis and polydactyly, with both inter- and intra-familial variability. JSRD are classified in six phenotypic subgroups: Pure JS; JS with ocular defect; JS with renal defect; JS with oculorenal defects; JS with hepatic defect; JS with orofaciodigital defects. With the exception of rare X-linked recessive cases, JSRD follow autosomal recessive inheritance and are genetically heterogeneous. Ten causative genes have been identified to date, all encoding for proteins of the primary cilium or the centrosome, making JSRD part of an expanding group of diseases called "ciliopathies". Mutational analysis of causative genes is available in few laboratories worldwide on a diagnostic or research basis. Differential diagnosis must consider in particular the other ciliopathies (such as nephronophthisis and Senior-Loken syndrome), distinct cerebellar and brainstem congenital defects and disorders with cerebro-oculo-renal manifestations. Recurrence risk is 25% in most families, although X-linked inheritance should also be considered. The identification of the molecular defect in couples at risk allows early prenatal genetic testing, whereas fetal brain neuroimaging may remain uninformative until the end of the second trimester of pregnancy. Detection of the MTS should be followed by a diagnostic protocol to assess multiorgan involvement. Optimal management requires a multidisciplinary approach, with particular attention to respiratory and feeding problems in neonates and infants. Cognitive and behavioral assessments are also recommended to provide young patients with adequate neuropsychological support and rehabilitation. After the first months of life, global prognosis varies considerably among JSRD subgroups, depending on the extent and severity of organ involvement.
Figures
Similar articles
-
Diagnosis of Joubert syndrome via ultrasonography.J Med Ultrason (2001). 2017 Apr;44(2):197-202. doi: 10.1007/s10396-016-0751-8. Epub 2016 Oct 26. J Med Ultrason (2001). 2017. PMID: 27785575
-
Joubert syndrome and related disorders.Handb Clin Neurol. 2013;113:1879-88. doi: 10.1016/B978-0-444-59565-2.00058-7. Handb Clin Neurol. 2013. PMID: 23622411 Review.
-
Clinical and molecular features of Joubert syndrome and related disorders.Am J Med Genet C Semin Med Genet. 2009 Nov 15;151C(4):326-40. doi: 10.1002/ajmg.c.30229. Am J Med Genet C Semin Med Genet. 2009. PMID: 19876931 Free PMC article. Review.
-
Orocraniofacial findings of a Pediatric Patient with Joubert Syndrome.Int J Clin Pediatr Dent. 2016 Oct-Dec;9(4):379-383. doi: 10.5005/jp-journals-10005-1394. Epub 2016 Dec 5. Int J Clin Pediatr Dent. 2016. PMID: 28127172 Free PMC article.
-
CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders.Am J Hum Genet. 2007 Jul;81(1):104-13. doi: 10.1086/519026. Epub 2007 May 18. Am J Hum Genet. 2007. PMID: 17564967 Free PMC article.
Cited by
-
Joubert syndrome presenting with motor delay and oculomotor apraxia.Case Rep Pediatr. 2011;2011:262641. doi: 10.1155/2011/262641. Epub 2012 Jan 26. Case Rep Pediatr. 2011. PMID: 22606509 Free PMC article.
-
Investigating embryonic expression patterns and evolution of AHI1 and CEP290 genes, implicated in Joubert syndrome.PLoS One. 2012;7(9):e44975. doi: 10.1371/journal.pone.0044975. Epub 2012 Sep 24. PLoS One. 2012. PMID: 23028714 Free PMC article.
-
Molar Tooth Sign with Deranged Liver Function Tests: An Indian Case with COACH Syndrome.Case Rep Pediatr. 2015;2015:385910. doi: 10.1155/2015/385910. Epub 2015 May 17. Case Rep Pediatr. 2015. PMID: 26075130 Free PMC article.
-
Joubert syndrome: the molar tooth sign of the mid-brain.Ann Med Health Sci Res. 2013 Apr;3(2):291-4. doi: 10.4103/2141-9248.113686. Ann Med Health Sci Res. 2013. PMID: 23919210 Free PMC article.
-
Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation.Nat Neurosci. 2013 Aug;16(8):1000-7. doi: 10.1038/nn.3451. Epub 2013 Jun 30. Nat Neurosci. 2013. PMID: 23817546 Free PMC article.
References
-
- Joubert M, Eisenring JJ, Andermann F. Familial dysgenesis of the vermis: a syndrome of hyperventilation, abnormal eye movements and retardation. Neurology. 1968;18:302–303. - PubMed
-
- Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, Hove MT, Fennell EB, Booth-Jones M, Ringdahl DM, Yachnis AT, Creel G, Frerking B. "Joubert syndrome" revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12:423–430. doi: 10.1177/088307389701200703. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
