

Clinical aspects of short-chain acyl-CoA dehydrogenase deficiency
- PMID: 20429031
- PMCID: PMC2946545
- DOI: 10.1007/s10545-010-9080-z
Clinical aspects of short-chain acyl-CoA dehydrogenase deficiency
Abstract
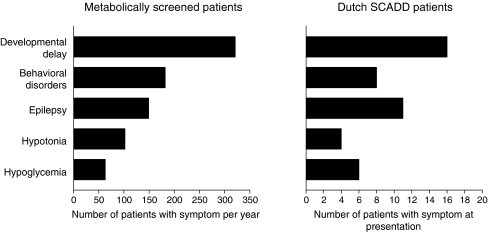
Short-chain acyl-CoA dehydrogenase deficiency (SCADD) is an autosomal recessive inborn error of mitochondrial fatty acid oxidation. SCADD is biochemically characterized by increased C4-carnitine in plasma and ethylmalonic acid in urine. The diagnosis of SCADD is confirmed by DNA analysis showing SCAD gene mutations and/or variants. SCAD gene variants are present in homozygous form in approximately 6% of the general population and considered to confer susceptibility to development of clinical disease. Clinically, SCADD generally appears to present early in life and to be most frequently associated with developmental delay, hypotonia, epilepsy, behavioral disorders, and hypoglycemia. However, these symptoms often ameliorate and even disappear spontaneously during follow-up and were found to be unrelated to the SCAD genotype. In addition, in some cases, symptoms initially attributed to SCADD could later be explained by other causes. Finally, SCADD relatives of SCADD patients as well as almost all SCADD individuals diagnosed by neonatal screening remained asymptomatic during follow-up. This potential lack of clinical consequences of SCADD has several implications. First, the diagnosis of SCADD should never preclude extension of the diagnostic workup for other potential causes of the observed symptoms. Second, patients and parents should be clearly informed about the potential lack of relevance of the disorder to avoid unfounded anxiety. Furthermore, to date, SCADD is not an optimal candidate for inclusion in newborn screening programs. More studies are needed to fully establish the relevance of SCADD and solve the question as to whether SCADD is involved in a multifactorial disease or represents a nondisease.
Figures
Similar articles
-
Biochemical, molecular, and clinical characteristics of children with short chain acyl-CoA dehydrogenase deficiency detected by newborn screening in California.Mol Genet Metab. 2012 May;106(1):55-61. doi: 10.1016/j.ymgme.2012.02.007. Epub 2012 Feb 9. Mol Genet Metab. 2012. PMID: 22424739
-
Clinical, biochemical, and genetic heterogeneity in short-chain acyl-coenzyme A dehydrogenase deficiency.JAMA. 2006 Aug 23;296(8):943-52. doi: 10.1001/jama.296.8.943. JAMA. 2006. PMID: 16926354
-
An unusually high frequency of SCAD deficiency caused by two pathogenic variants in the ACADS gene and its relationship to the ethnic structure in Slovakia.BMC Med Genet. 2018 Apr 20;19(1):64. doi: 10.1186/s12881-018-0566-0. BMC Med Genet. 2018. PMID: 29678161 Free PMC article.
-
Newborn screening for disorders of fatty-acid oxidation: experience and recommendations from an expert meeting.J Inherit Metab Dis. 2010 Oct;33(5):521-6. doi: 10.1007/s10545-010-9076-8. Epub 2010 Apr 7. J Inherit Metab Dis. 2010. PMID: 20373143 Review.
-
Short-chain acyl-CoA dehydrogenase deficiency: from gene to cell pathology and possible disease mechanisms.J Inherit Metab Dis. 2017 Sep;40(5):641-655. doi: 10.1007/s10545-017-0047-1. Epub 2017 May 17. J Inherit Metab Dis. 2017. PMID: 28516284 Review.
Cited by
-
[Newborn screening as a predictive genetic test: principles and challenges].Wien Med Wochenschr. 2012 Apr;162(7-8):168-75. doi: 10.1007/s10354-012-0062-7. Epub 2012 Mar 28. Wien Med Wochenschr. 2012. PMID: 22451084 German.
-
Down-regulation of metabolic proteins in hepatocellular carcinoma with portal vein thrombosis.Clin Proteomics. 2017 Aug 2;14:29. doi: 10.1186/s12014-017-9164-y. eCollection 2017. Clin Proteomics. 2017. PMID: 28785178 Free PMC article.
-
Compound heterozygous mutations of ACADS gene in newborn with short chain acyl-CoA dehydrogenase deficiency: case report and literatures review.Korean J Pediatr. 2016 Nov;59(Suppl 1):S45-S48. doi: 10.3345/kjp.2016.59.11.S45. Epub 2016 Nov 30. Korean J Pediatr. 2016. PMID: 28018444 Free PMC article.
-
Stable isotope labeling by essential nutrients in cell culture for preparation of labeled coenzyme A and its thioesters.Anal Chem. 2011 Feb 15;83(4):1363-9. doi: 10.1021/ac1027353. Epub 2011 Jan 26. Anal Chem. 2011. PMID: 21268609 Free PMC article.
-
Newborn Screening for Lysosomal Storage Disorders: Views of Genetic Healthcare Providers.J Genet Couns. 2016 Apr;25(2):373-84. doi: 10.1007/s10897-015-9879-8. Epub 2015 Aug 29. J Genet Couns. 2016. PMID: 26315880
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
