

Phenotype and genotype characterization of adenine phosphoribosyltransferase deficiency
- PMID: 20150536
- PMCID: PMC2844298
- DOI: 10.1681/ASN.2009080808
Phenotype and genotype characterization of adenine phosphoribosyltransferase deficiency
Abstract
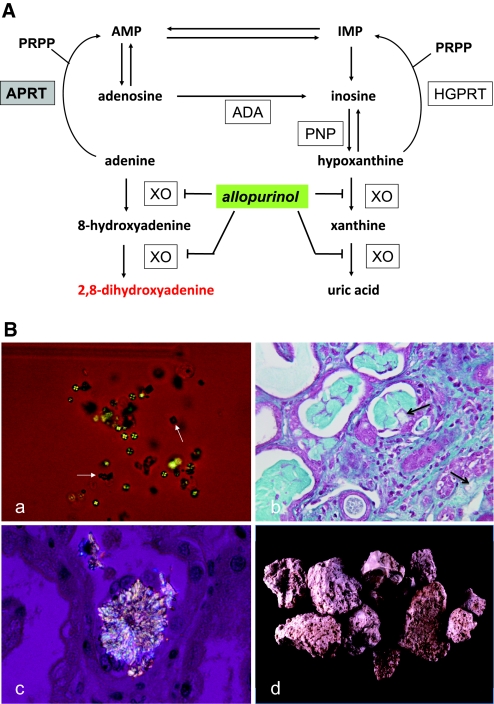
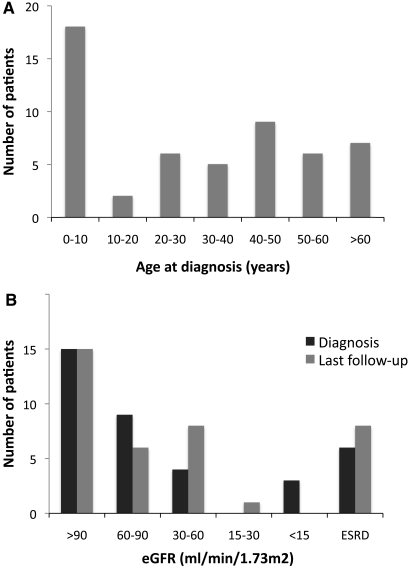
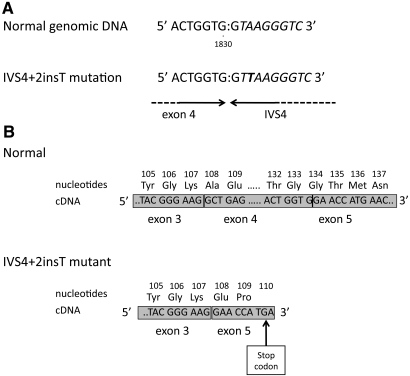
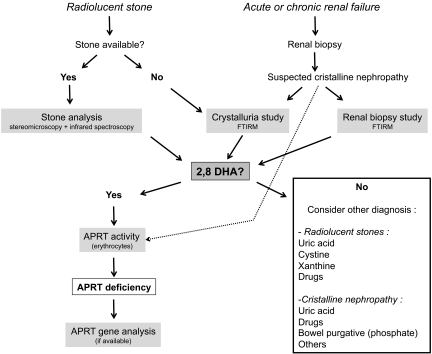
Adenine phosphoribosyltransferase (APRT) deficiency is a rare autosomal recessive disorder causing 2,8-dihydroxyadenine stones and renal failure secondary to intratubular crystalline precipitation. Little is known regarding the clinical presentation of APRT deficiency, especially in the white population. We retrospectively reviewed all 53 cases of APRT deficiency (from 43 families) identified at a single institution between 1978 and 2009. The median age at diagnosis was 36.3 years (range 0.5 to 78.0 years). In many patients, a several-year delay separated the onset of symptoms and diagnosis. Of the 40 patients from 33 families with full clinical data available, 14 (35%) had decreased renal function at diagnosis. Diagnosis occurred in six (15%) patients after reaching ESRD, with five diagnoses made at the time of disease recurrence in a renal allograft. Eight (20%) patients reached ESRD during a median follow-up of 74 months. Thirty-one families underwent APRT sequencing, which identified 54 (87%) mutant alleles on the 62 chromosomes analyzed. We identified 18 distinct mutations. A single T insertion in a splice donor site in intron 4 (IVS4 + 2insT), which produces a truncated protein, accounted for 40.3% of the mutations. We detected the IVS4 + 2insT mutation in two (0.98%) of 204 chromosomes of healthy newborns. This report, which is the largest published series of APRT deficiency to date, highlights the underdiagnosis and potential severity of this disease. Early diagnosis is crucial for initiation of effective treatment with allopurinol and for prevention of renal complications.
Figures
Similar articles
-
Long-term renal outcomes of APRT deficiency presenting in childhood.Pediatr Nephrol. 2019 Mar;34(3):435-442. doi: 10.1007/s00467-018-4109-x. Epub 2018 Nov 15. Pediatr Nephrol. 2019. PMID: 30443743 Free PMC article.
-
Adenine phosphoribosyltransferase deficiency in children.Pediatr Nephrol. 2012 Apr;27(4):571-9. doi: 10.1007/s00467-011-2037-0. Epub 2012 Jan 3. Pediatr Nephrol. 2012. PMID: 22212387
-
Two families with compound heterozygosity for adenine phosphoribosyltransferase deficiency.Pediatr Nephrol. 2010 Jun;25(6):1173-6. doi: 10.1007/s00467-009-1430-4. Epub 2010 Jan 26. Pediatr Nephrol. 2010. PMID: 20101413
-
[2,8-dihydroxyadenine urolithiasis: case report and literature review].Urologia. 2011 Oct-Dec;78(4):305-9. doi: 10.5301/RU.2011.8307. Urologia. 2011. PMID: 21553389 Review. Italian.
-
Adenine phosphoribosyltransferase deficiency.Clin J Am Soc Nephrol. 2012 Sep;7(9):1521-7. doi: 10.2215/CJN.02320312. Epub 2012 Jun 14. Clin J Am Soc Nephrol. 2012. PMID: 22700886 Review.
Cited by
-
Quantitative UPLC-MS/MS assay of urinary 2,8-dihydroxyadenine for diagnosis and management of adenine phosphoribosyltransferase deficiency.J Chromatogr B Analyt Technol Biomed Life Sci. 2016 Nov 15;1036-1037:170-177. doi: 10.1016/j.jchromb.2016.09.018. Epub 2016 Sep 14. J Chromatogr B Analyt Technol Biomed Life Sci. 2016. PMID: 27770717 Free PMC article.
-
Successful Management of Adenine Phosphoribosyl Transferase Enzyme Deficiency in Renal Transplantation: A Case Report.Indian J Nephrol. 2024 Jul-Aug;34(4):383-385. doi: 10.25259/IJN_167_2024. Epub 2024 Jun 17. Indian J Nephrol. 2024. PMID: 39156833 Free PMC article.
-
Long-term renal outcomes of APRT deficiency presenting in childhood.Pediatr Nephrol. 2019 Mar;34(3):435-442. doi: 10.1007/s00467-018-4109-x. Epub 2018 Nov 15. Pediatr Nephrol. 2019. PMID: 30443743 Free PMC article.
-
The genetics of kidney stone disease and nephrocalcinosis.Nat Rev Nephrol. 2022 Apr;18(4):224-240. doi: 10.1038/s41581-021-00513-4. Epub 2021 Dec 14. Nat Rev Nephrol. 2022. PMID: 34907378 Review.
-
Rapidly progressive kidney dysfunction and crystal casts associated with adenine phosphoribosyltransferase (APRT) deficiency-lessons for the clinical nephrologist.J Nephrol. 2021 Dec;34(6):2147-2149. doi: 10.1007/s40620-021-01042-w. Epub 2021 Apr 7. J Nephrol. 2021. PMID: 33826114 No abstract available.
References
-
- Cartier P, Hamet M: Purine phosphoribosyltransferase activity of human erythrocytes. Technique of determination [in French]. Clin Chim Acta 20: 205– 214, 1968 - PubMed
-
- Sahota AS, Tischfield JA, Kamatani N, Simmonds HA: Adenine phosporibosyltransferase deficiency and 2,8-dihydroxyadenine lithiasis. In: The Metabolic and Molecular Bases of Inherited Disease, 8th Ed., edited by Scriver CR, Baudet AL, Sly WS, Valle D. New York, McGraw-Hill Division, 2001, pp. 2571– 2584
-
- Cartier P, Hamet M: The normal metabolism of uric acid. Adv Nephrol Necker Hosp 3: 3– 28, 1974 - PubMed
-
- Hesse A, Miersch WD, Classen A, Thon A, Doppler W: 2,8-Dihydroxyadeninuria: Laboratory diagnosis and therapy control. Urol Int 43: 174– 178, 1988 - PubMed
-
- Benedetto B, Madden R, Kurbanov A, Braden G, Freeman J, Lipkowitz GS: Adenine phosphoribosyltransferase deficiency and renal allograft dysfunction. Am J Kidney Dis 37: E37, 2001 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
